Inorganic Builder Plugin, Version 0.1
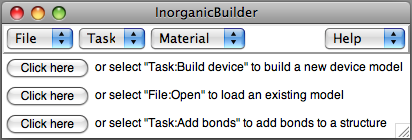 The InorganicBuilder plugin provides tools for many of the functions
needed to set up simulations of molecules and inorganic structures. The
functions of the plugin are organized into several Tasks. Initially, the plugin displays buttons for three common activities, which correspond to options in the Task and File menues.
The InorganicBuilder plugin provides tools for many of the functions
needed to set up simulations of molecules and inorganic structures. The
functions of the plugin are organized into several Tasks. Initially, the plugin displays buttons for three common activities, which correspond to options in the Task and File menues.
Menus
The plugin contains four top-level menus, File, Task, Material, and Help.File Menu
The File menu has 2 options, Open and Save as. Save as saves the state of the plugin, including specifications for the last-built device and any modifications to the Material library. The Open option restores a previously saved state file. Note that it does not load any previously-generated structures into VMD. Those must be loaded separately.Task Menu
The Task menu provides tools for performing several common operations needed to build an atomic model of an inorganic device. These operations are explained below.Build device
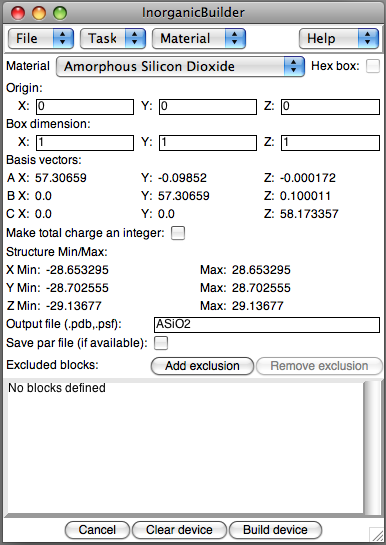 This task lets you set up the basic parameters, including material and
dimensions, for the initial device structure. The following values
describe the structure:
This task lets you set up the basic parameters, including material and
dimensions, for the initial device structure. The following values
describe the structure:
- Material: Select a material to specify the corresponding unit cell and basis cell dimensions. Additional materials can be added to this menu using the text interface.
- Hex box: For materials with hexagonal symmetry in the X-Y plane, checking this box will convert the structure to a hexagonal form.
- Origin: Specify the coordinates of the center of the device box
- Box dimension: Three integers specify the number of unit cells in the A, B, and C directions. When using hexagonal symmetry, Diameter defines the diameter of the inscribed cylinder, and height defines the number of unit cells in the Z dimension.
- Make total charge an integer: If the total charge of the system is not an integer, the charge of all the atoms is scaled slightly to make the total system charge an integer. Then the total charge is recomputed. Due to insufficient precision in the PDB file format, there may still be a very small charge correction necessary, so any remaining charge is added to the first atom in the PDB file.
- Output file: The file name to store the resulting PSF/PDB files
- Save par file: For some material types, the plugin includes a parameter file in the CHARMM format which may be appropriate for some simulations. When available, this file will be saved along with the PDB and PSF files.
Clicking Draw Box after specifiying the box parameters will construct a wire frame outline of the device (including any exclusions) in the VMD OpenGL window.
In addition, you may cut regions out of the unit cell before you construct it, by Adding exclusions, regions to remove from the structure before constructing the PSF/PDB files. Defined regions can be removed before construction by selecting one and clicking Remove exclusion.
To create a device of interest, after the block of material is specified, regions of various shapes are removed. Several different types of exclusions are available for constructing the device:
- Parallelopiped: Specify one corner of the parallelopiped, and the three vectors defining the three edges intersecting at that corner.
- Cylinder: Specify the centers of the bottom and the top of the cylinder, as well as the radius.
- Sphere: Specify the center and radius of the sphere.
- Cone: Specify the center of the base of the cone, the apex, and the radius of the base.
- Tetrahedron: Specify one corner of the tetrahedron, and the three vectors defining the three edges intersecting at that corner.
- VMD selection: Specify a VMD selection (using the syntax of the atomselect command) to either include or exclude. Selections will be applied in the order that they are added to the exclusion list. VMD selection exclusions will not show up when the Draw Box button is used, since they must be applied while the structure is being filled with atoms.
After defining the device size and all desired exclusions, press the
Build structure button to construct the model. Clicking
Cancel closes the plugin window.
Add bonds
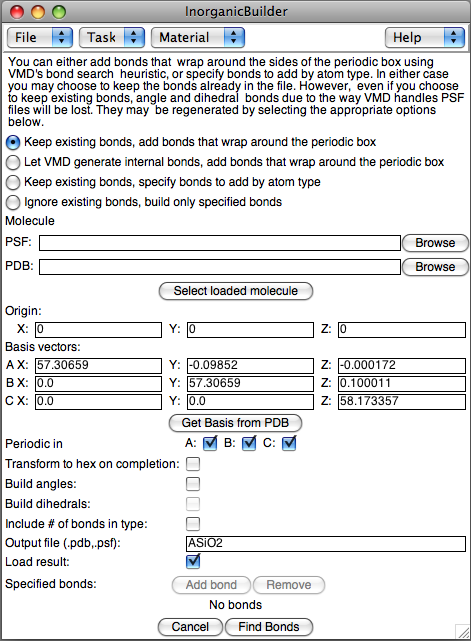 When reading in a structure with no bond information, VMD uses a
distance-based algorithm to guess the bonds. This algorithm works well
for many models, but does not take into account bonds that wrap around
the walls of a periodic cell, or bond lengths in some materials, especially amorphous materials. This task enhances the built-in VMD bond
search algorithm by letting you add bonds to a previously-generated structure. The first prompt allows you to select one of the four available methods for generating bonds:
When reading in a structure with no bond information, VMD uses a
distance-based algorithm to guess the bonds. This algorithm works well
for many models, but does not take into account bonds that wrap around
the walls of a periodic cell, or bond lengths in some materials, especially amorphous materials. This task enhances the built-in VMD bond
search algorithm by letting you add bonds to a previously-generated structure. The first prompt allows you to select one of the four available methods for generating bonds:
- Keep all existing bonds already defined in VMD, add bonds that wrap around the periodic box
- Let VMD regenerate internal bonds, add bonds that wrap around the periodic box
- Keep existing bonds, specify bonds to add by atom type
- Ingnore existing bonds, and create a PDB/PSF file containing only specified bonds
- Molecule: Select a structure, either by selecting a molecule already loaded in VMD, or by selecting the PSF and PDB files for a stored structure.
- Origin and Basis: If you just created this structure, the Origin and Basis parameters will be carried over from the Build Crystal task, otherwise, enter the Origin (center of the box) and the basis vectors for the periodic box.
- Periodic in: Only generate bonds wrapping around the basis cell for the specified directions.
- Transform to hex: If the unit cell has hexagonal symmetry in the X-Y plane, transform to a hexagonal box.
- Build angles/dihedrals: After building the two-atom bonds, also generate angle and dihedral bonds
- Include # of bonds: Rename the atom types so that the last digit of each atom's type includes the number of bonds as the last character. For instance, a silicon atom with 4 bonds would have the atom type "SI4"
- Output file: The name of the PSF/PDB files of the structure with bonds
- Load result: After generating and storing the structure, load it in to VMD
Specify what element types should be bonded by pressing the Add bond button. Enter the two element names and the maximum radius for the bond. The Remove button can be used to get rid of unwanted bond types.
After defining these parameters, press the Find Bonds button
to construct the model. Clicking Cancel closes the plugin window.
Find surface atoms
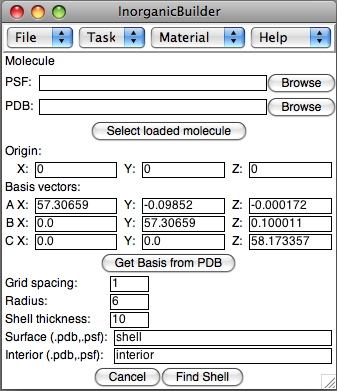 This task allows a device to be separated into two sets of files, one
containing atoms within a certain distance of the surface, and the other
containing the interior atoms. The shell of surface atoms is defined by
its proximity to vacuum. A grid is placed over the volume of the
periodic cell, and each grid point is considered a vacuum grid point if
no atoms fall within a sphere (bubble) of radius r centered on that grid
point. Then, surface atom shell of the desired thickness t is determined
by selecting all atoms closer than r + t of a vacuum grid point. These
parameters are entered into the plugin as follows:
This task allows a device to be separated into two sets of files, one
containing atoms within a certain distance of the surface, and the other
containing the interior atoms. The shell of surface atoms is defined by
its proximity to vacuum. A grid is placed over the volume of the
periodic cell, and each grid point is considered a vacuum grid point if
no atoms fall within a sphere (bubble) of radius r centered on that grid
point. Then, surface atom shell of the desired thickness t is determined
by selecting all atoms closer than r + t of a vacuum grid point. These
parameters are entered into the plugin as follows:
- Molecule: Select a structure, either by selecting a molecule already loaded in VMD, or by selecting the PSF and PDB files for a stored structure.
- Origin and Basis: If you just created this structure, the Origin and Basis parameters will be carried over from the Build Crystal task, otherwise, enter the Origin (center of the box) and the basis vectors for the periodic box.
- Grid spacing: The approximate resolution of the grid for deteriming the vacuum region. This is only an approximate parameter, since the actual distance is adjusted in each direction so that there are an integer number of grid points along each basis vector.
- Radius: A grid point is considered a vacuum if no atoms lie within the sphere of the specified radius around the grid point. Alternatively, atoms can be viewed as spheres of this radius, and a grid point is only a vacuum if it lies outside all the atoms.
- Shell thickness: The resulting surface shell will contain atoms within this distance of the surface.
- Surface: The PSF/PDB files for the surface shell
- Interior: The PSF/PDB files for the interior atoms
Solvate box
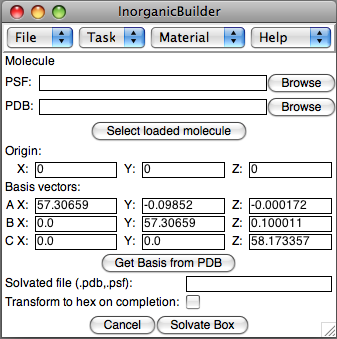 The Solvate task uses VMD's Solvate plugin to solvate a system according to the specified basis vectors.
The Solvate task uses VMD's Solvate plugin to solvate a system according to the specified basis vectors.
- Molecule: Select a structure, either by selecting a molecule already loaded in VMD, or by selecting the PSF and PDB files for a stored structure.
- Origin and Basis: If you just created this structure, the Origin and Basis parameters will be carried over from the Build Crystal task, otherwise, enter the Origin (center of the box) and the basis vectors for the periodic box.
- Solvated file: The PSF/PDB files for the resulting solvated structure
- Transform to hex: If the basis vectors permit it, the structure will be transformed into a version that is hexagonal in the X-Y plane.
Material Menu
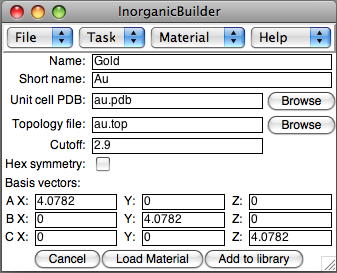 The plugin lets you define new materials to use for constructing devices. The materials can be defined as part of your InorganicBuilder session, and saved for reloading into another session. Materials currently in the library may also be viewed.
The plugin lets you define new materials to use for constructing devices. The materials can be defined as part of your InorganicBuilder session, and saved for reloading into another session. Materials currently in the library may also be viewed.
To add a new material, the plugin requires several pieces of information.
- Name and Short name: The Name is a longer, more descriptive name for the material. The Short Name is a shorthand name that should be a unique identifier for the material.
- Unit coll PDB: A PDB file containing names and coordinates of the atoms in the unit cell.
- Topology file: An optional file describing the unit-cell atom types in a CHARMM-compatible format.
- Cutoff: The cutoff radius for periodic bond searches performed by the plugin. This does not affect VMD's internal bond search routine, or the plugin's specify-bonds algorithm.
- Hex symmetry: Specifies whether the unit cell has the correct basis vectors to be represented as a hexagonal cell in the X-Y plane.
- Basis vectors: The basis vectors for the unit cell.


