Dowser Plugin, Version 1.1
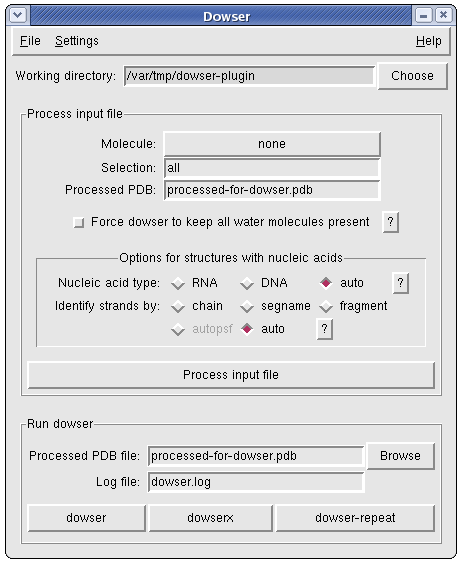
The Dowser plugin can be used to process a pdb file for use with Dowser, a program that finds cavities in proteins and solvates them according to an energy criterium. You will need to install Dowser yourself. Please refer to the Dowser manual. In the instructions you will find that you need to source 'dowserinit' before running dowser.
The main reason for the existence of this plugin is that Dowser has been extended to work with nucleic acids, and there's a considerable amount of processing that has to be done in this case. In order to have Dowser to work with nucleic acids, you will need to use additional dictionary definitions. For DNA, please refer to Cameron Mura's website. For RNA, please refer to this page.
Note that this is a preliminary version and the development of this plugin is in progress. Please cite the following reference in any publication that uses the Dowser plugin:
Existing water molecules
Dowser identifies existing water molecules (which it calls crystallographic water molecules) as HETATM entries atom name "O" and residue name "HOH". While processing the input file, the Dowser plugin converts all waters to follow this convention.
Dowser has the option of testing crystal water energies to decide if they should be kept or not (default), not test crystal water energies (-noxtalwater), or only test them (-onlyxtalwater). See the Dowser section below for more details.
The plugin offers an option to force Dowser to keep all water molecules initially present in the structure. It uses psfgen to add as many hydrogens as needed and sets the residue name of all water molecules to TRE. The Dowser's parameters for this residues are exactly the same as for HOH, except that Dowser will not test the position of these water molecules, regardless of the command-line options for crystallographic waters described above.
Processing structures containing nucleic acids
Setting the nucleic acid type
Nucleic acid residue names must be converted, and the convertion involved determining if the nucleic acid is DNA or RNA. If the nucleic acid type is not given, the plugin tries to identify the type on a residue-basis. This means that if VMD residue assignment is not correct due to some distortions in the structure, processing of nucleic acid residues will fail. If you provide a psf file, residue assignment by VMD should be very reliable and the plugin should have to problems. In any case, unless your structure contains both DNA and RNA, it is recommended that you specify the nucleic acid type.
Processing nucleic acid termini
In order to correctly process the terminal residues of each nucleic acid strand, the plugin first needs to identify the strands. The currently available methods to do this are:
chain: uses the chain identification in the PDB; to use this method, make sure each nucleic acid strand has a different chain identifier (recommended)
segname: uses the segname identification in the PDB; to use this method, make sure each nucleic acid strand has a different segname identifier (recommended)
fragment: uses fragment information derived by VMD when analysing the structure
autopsf: uses the same algorithm used by the AutoPSF plugin to split chains (not implemented yet)
auto: the auto option uses "fragment" if a PSF file was given or "autopsf" otherwise (not implemented yet, defaults to "fragment" always as of now)
Regardless of the method used, you should check if the termini have been correctly identified when this information is presented to you. If there is a problem, your best bet is to make sure each strand has a different chain identifier and use the "chain" method.
Currently, only uncharged termini (without a terminal phosphate group) are supported, both for DNA and RNA. Terminal phosphate groups are silently ignored.
Dowser options

Dowser options can be chosen through Settings->Dowser options...
Include hetero atoms (-hetero): hetero atoms (HETATM records in the PDB file) will be included, with the exception of crystallographic water molecules identified as having atom name "O" and residue name "HOH". (The default is to use only ATOM records.) Note that all HETATM records are converted to ATOM by VMD, so if you do not want these atoms to be included, delete them from the input PDB before you start using the Dowser plugin.
For a description of the other options, please refer to the Dowser manual.
Combining waters
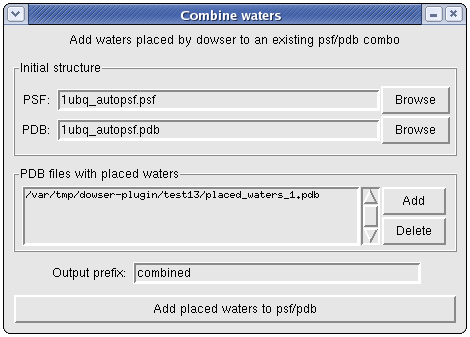
If you have a psf/pdb combo and wish to add the waters placed by dowser, you can do so by clicking File->Combine waters... The plugin keeps track of all waters placed by Dowser in the current session. Thus you can, for example, run dowserx followed by a series of runs of dowser-repeat, and then combine the waters placed in all runs at once.


