NAMD and VMD at SC2002
Gordon Bell Award Finalist NAMD Paper
Title:
NAMD: Biomolecular Simulation on Thousands of Processors
Location:
Room 309-310
Time:
2:30 Wednesday
Authors:
James C. Phillips, Gengbin Zheng, Sameer Kumar, Laxmikant V. Kale
Abstract:
NAMD is a fully featured, production molecular dynamics program for high performance simulation
of large biomolecular systems. We have previously, at SC2000, presented scaling results for
simulations with cutoff electrostatics on up to 2048 processors of the ASCI Red machine,
achieved with an object-based hybrid force and spatial decomposition scheme and an aggressive
measurement-based predictive load balancing framework. We extend this work by demonstrating
similar scaling on the much faster processors of the PSC Lemieux Alpha cluster, and for
simulations employing efficient (order N log N) particle mesh Ewald full electrostatics. This
unprecedented scalability in a biomolecular simulation code has been attained through latency
tolerance, adaptation to multiprocessor nodes, and the direct use of the Quadrics Elan library in
place of MPI by the Charm++/Converse parallel runtime system.
Theoretical Biophysics (TBG) and NCSA Itanium 2 Cluster and Display Wall Demo and Talk
Title:
NAMD and VMD: Live Molecular Dynamics Simulation and Visualization
on NCSA Itanium Linux Cluster and Tiled Display Wall
Location:
NCSA booth
Times:
8pm Monday, 1pm Tuesday
Investigators:
Klaus Schulten, Robert Skeel, Laxmikant Kale
Developers:
James Phillips, John Stone, Justin Gullingsrud, Gengbin Zheng, Paul Grayson (all TBG)
Presenter:
Jim Phillips, TBG
Contacts:
Bill Bell, Bruce Loftis, and Rob Pennington at NCSA
TBG Support:
Update display wall port of VMD, provide and present demo simulation.
What will be demonstrated:
A NAMD simulation of GlpF (see below)
will be running on the NCSA Itanium 2 Linux cluster at SC2002
VMD will be demonstrated on NCSA's tiled display wall
powered by a cluster of low-cost graphics workstations
built with commodity parts. VMD will be used to visualize
the running simulation.
The process of tuning NAMD for the Itanium processor will be
discussed, and serial and parallel performance results will be presented.
NAMD and VMD are developed by the Theoretical Biophysics Group at the
University of Illinois. NAMD and VMD development is supported by
the NIH National Center for Research Resources
and the National Science Foundation.
Theoretical Biophysics (TBG) and Sun Microsystems XVR-4000 Demo
Title:
NAMD and VMD: Interactive Molecular Dynamics Simulation
Location:
Sun Microsystems booth
Times:
various, unknown
Investigators:
Klaus Schulten, Robert Skeel, Laxmikant Kale
Developers:
James Phillips, John Stone, Justin Gullingsrud, Gengbin Zheng, Paul Grayson (all TBG)
Presenter:
Sun personnel
Contacts:
Travis Bryson at Sun Microsystems
TBG Support:
Technical advice, NAMD and VMD inputs for demo simulation and display.
What will be demonstrated:
A simulation of GlpF (see below) will be performed using
NAMD on a 20
processor SunFire 6800 with ClusterTools 4.0 MPI parallel communication
software. VMD
will be running on the same machine and displayed by
XVR-4000 graphics boards in two SunFire V880z systems. The V880z
systems are connected to the 6800 via a high bandwidth multiple Myrinet
connection.
NAMD and VMD are developed by the Theoretical Biophysics Group at the
University of Illinois. NAMD and VMD development is supported by
the NIH National Center for Research Resources
and the National Science Foundation.
About the GlpF Demo Simulation (In Both Sun and NCSA Booths)
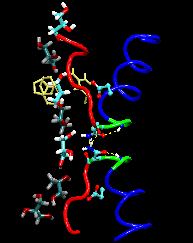
Aquaporins are channel proteins abundantly present in all life forms,
for example, bacteria, plants, and in the kidneys, the eyes, and the
brain of humans. These proteins conduct water and small molecules, but
no ions, across the cell walls. Their defective forms are known to
cause diseases, e.g., diabetes insipidus, or cataracts. The
molecular modeling program, NAMD, along
with large parallel computers at the Pittsburgh and Illinois supercomputing centers
permitted researchers now to model aquaporins in the natural
environment of membrane and water in one of the largest molecular
dynamics simulations ever (over 100,000 atoms). The simulations
revealed in unprecedented detail how cells conduct water and glycerol, a molecule that serves cells' metabolism. The
simulations provided a movie of the entire conduction process.
A greatly reduced model of a single GlpF channel, consisting of only
4210 atoms, 3295 of them fixed, will be used to demonstrate the power
of NAMD to accelerate even small simulations on modest numbers of processors.
The Tcl scripting capabilities of NAMD are used to apply a constant force,
driving a single glycerol molecule back and forth through the channel.
While the force biases the movement of the glycerol, it still takes over
100 ps of dynamics to find the proper alignment to pass through the
specificity filter region of this remarkable molecule.


