VMD 1.9.3
The Theoretical and Computational Biophysics Group is pleased to announce VMD version 1.9.3.VMD incorporates many new improvements for high quality rendering and export of molecular scenes, new analysis features, support for new molecular data file formats, and many performance improvements. Many new and updated structure building and analysis tools have been added in this release, easing the process of preparing, running, and analyzing biomolecular simulations. This release also contains many performance and efficiency improvements that are particiularly beneficial for modeling, visualizing, and analyzing very large structures with over 100 million atoms. VMD makes extensive use of multi-core processors, vector instruction set extensions such as Intel SSE, AVX, and AVX-512, and IBM VSX, as well as GPU acceleration to speed up computationally demanding analysis and visualization tasks including key structure and trajectory analysis features, interactive molecular dynamics, and high-quality ray tracing of molecular scenes.
- VMD 1.9.3 Documentation, Release Notes, Tutorials
- Download VMD 1.9.3 for MacOS X, Unix, or Windows
- VMD 1.9.3 Development and Release History (large)
Major features included in VMD 1.9.3:
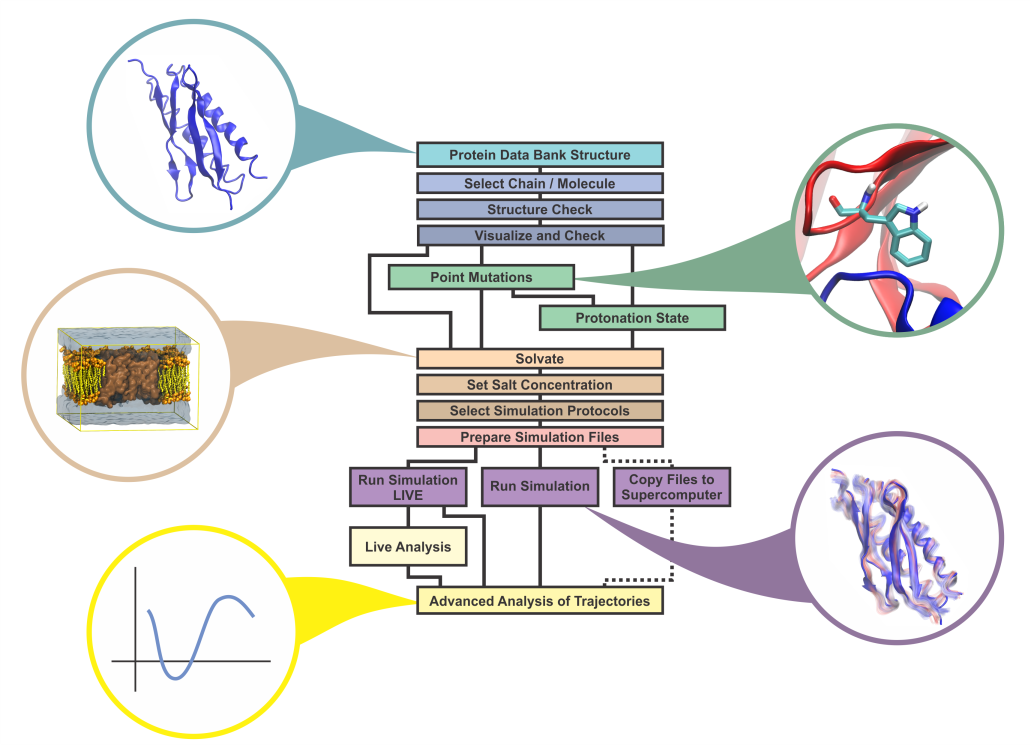
Connecting the user-friendly molecular graphics program VMD to the widely adopted MD program NAMD, the new QwikMD plugin guides users to prepare common molecular simulations in just a few minutes, allowing for quickly study of point mutations, partial deletions or even steering experiments. While making it easy for a new user to perform simulations, QwikMD also works as a learning tool with "info buttons" that guide the user and provide the theoretical background underlying the procedures that are followed. Each "info button" generates a window where the user can find fundamental information related to Molecular Dynamics simulations, such as the description of solvent models and protein structure, as well as short explanations of the options available in QwikMD graphical interface and the actions triggered by the buttons. For more information, the user is redirected to web-pages through links present in the bottom of the information window. QwikMD incorporates logging of procedures for reproducibility, and to enable simulation workflows and protocols to be shared among users. Documentation and tutorials are available on the plugin website, and the key attributes of the design and philosophy behind QwikMD are reported here.
The new QwikMD integrative modeling plugin assists with
preparation and analysis of molecular simulations,
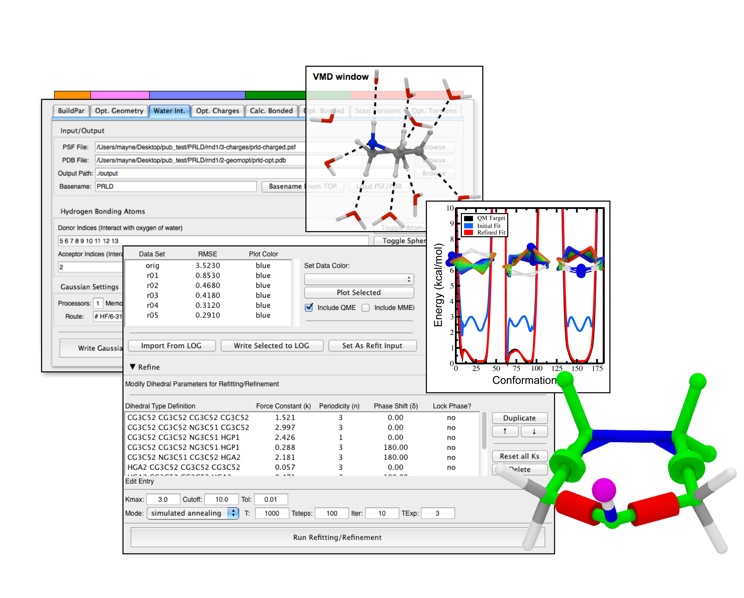
as shown in this "standard MD workflow" exampleThe Force Field Toolkit (ffTK) plugin provides a comprehensive toolset for the development of CHARMM-compatible (e.g., CGenFF) force field parameters, including charges, bonds, angles, and dihedrals. Two new major features have been added to the Force Field Toolkit for the VMD 1.9.3 release.
The latest Force Field Toolkit plugin assists with
development of CHARMM-compatible parametersA new tool has been added to the BuildPar toolset that processes output from the CGenFF Program webserver to automatically construct all of the structural and parameter input required for entry into the ffTK parameterization workflow, bypassing a common stumbling block in structure preparation. The CGenFF Program web server performs an analysis of molecular structure and assigns parameters based on analogy to molecules contained with in the standard CGenFF parameter set. ffTK can now leverage the output from this resource to automatically prepare the PSF+PDB files required by ffTK, analyze penalty scores associated with each returned parameter, visualize all instances of a given parameter directly within the molecular structure, and write a parameter file that is initialized to the CGenFF values as a starting point for refinement.
A new analysis and plotting utility, named Torsion Explorer (accessable from the Scan Torsions tab), is designed specifically to aid in troubleshooting difficult dihedral parameter fits providing visual and numerical analysis of the relaxed QM torsion scans. The key function of this tool is to load each optimized structural conformation into VMD and to construct a color plot of the associated potential energy surface (PES) for side-by-side comparison. Numerical analysis can also be performed to provide detailed information on how all dihedrals change throughout the scan, helping to identify highly coupled torsions that often complicate parameter fitting.
In addition to the major tools described above, ffTK contains several new but smaller features, such as the ability to output the MM relaxed conformational scan used as the baseline in dihedral fitting, and bug fixes. Documentation and screencast tutorials are available on the plugin website.
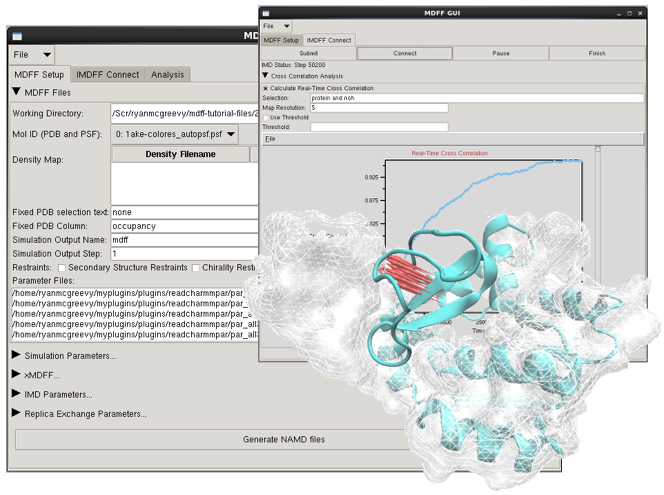
VMD 1.9.3 contains several updates for the Molecular Dynamics Flexible Fitting (MDFF) Method. The MDFF setup plugin now supports the use of multiple density maps in a single MDFF simulation. The MDFF graphical user interface (GUI) in the Modeling section of the VMD Extensions menu has been updated to reflect the use of multiple maps in addition to many other usability improvements. The cispeptide, chirality, and the new TorsionPlot, and QwikMD plugins now connect to the MDFF GUI to simplify finding and fixing errors or setting up your structure for MDFF. Both the MDFF GUI and command line plugins now use CHARMM36 as the default parameter set.
 Left: New MDFF graphical interfaces for analysis and interactive simulation.
Left: New MDFF graphical interfaces for analysis and interactive simulation.
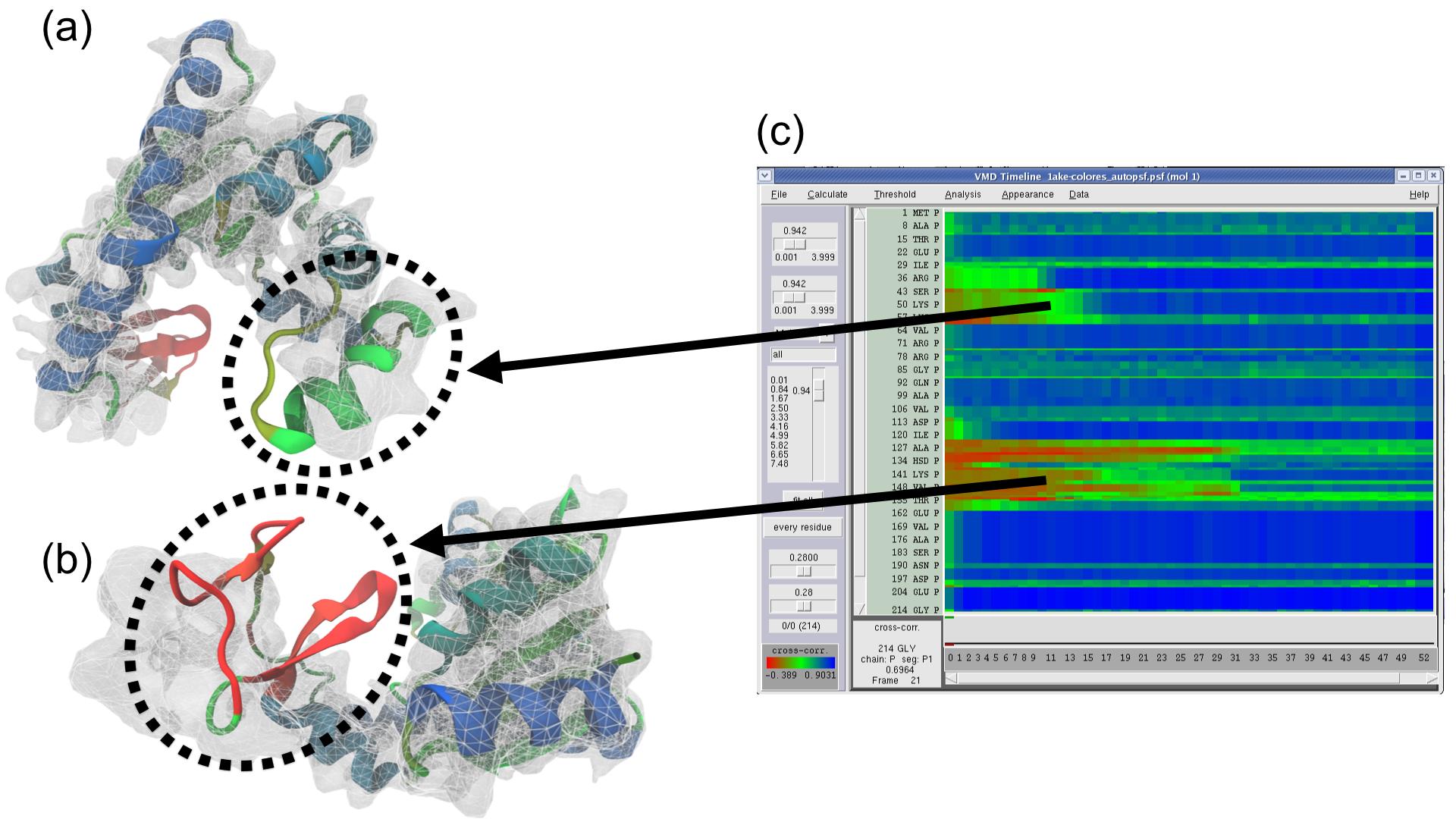
Right: GPU-accelerated MDFF hybrid structure quality-of-fit
cross correlation analysis shown in the Timeline plugin.The collective variables module implemented in VMD (and shared with NAMD and LAMMPS) incorporates many new features and improvements, including: histogram calculation on ensembles of variables, with optional weights; probability distribution restraints analysis; newly contributed variable types "dipoleAngle" (Alejandro Bernardin), "groupCoordNum" (Alan Grossfield); scripting command "cvcflags" to optimize performance of complex colvars; and improved error handling in user input and scripting.
VMD 1.9.3 contains many features for generating high quality renderings of molecular graphics and for creation of movies of both static structures and molecular dynamics simulation trajectories.
Proto-cell with ambient occlusion lighting, shadows, depth-of-field
focal blur, and reflections, rendered with the newest GPU-accelerated
TachyonL-OptiX parallel ray tracing engine built-into VMDVMD includes an improved built-in GPU-accelerated version of the Tachyon ray tracing engine, using NVIDIA CUDA and OptiX to achieve interactive ray tracing performance, even for molecular systems containing tens to hundreds of millions of particles. The improved GPU-accelerated "TachyonL-OptiX" ray tracer supports the vast majority of Tachyon rendering features, with emphasis on ambient occlusion lighting, shadows, depth-of-field, high quality transparent surface rendering, and parallel movie rendering for large complexes such as viruses, and cellular organelles.
The new GPU ray tracing engine in VMD 1.9.3 supports the generation of omnidirectional stereoscopic images and movies, suitable for use with YouTube VR movies, which are playable on a wide variety of VR headsets based on phones and computer-driven head mounted displays, including Google Cardboard, GearVR, Oculus Rift, and others. A new VMD VR movie rendering minitutorial details the steps necessary for creating VR movie content for YouTube and VR headsets.
VMD 1.9.3 also debuts a new built-in interactive CPU-based ray tracing engine based on Intel OSPRay, that allows systems based on Intel x86 CPUs and in particular the new Xeon Phi Knight's Landing processors to interactively display VMD scenes with ambient occlusion lighting, shadows, and depth-of-field, with support for parallel movie rendering. The new "TachyonL-OSPRay" renderer in VMD 1.9.3 achieves higher performance than the standard Tachyon renderer, and supports many of the most widely used VMD materials and shading features. The new OSPRay-based ray tracing engine in VMD is outlined in this recent article.
The updated ViewChangeRender plugin provides an easy-to-use graphical interface for managing multiple VMD camera viewpoints and making movies that fly the camera between multiple viewpoints. The latest version of ViewChangeRender also supports improved parallel movie rendering on clusters and supercomputers.
VMD molecular scenes with depth-of-field focal blur,
ambient occlusion lighting, and shadows, rendered with the
interactive ray tracing engines built-into VMDThe speed of the new interactive ray tracing engines in VMD 1.9.3 allow users to interactively visualize moderate size molecular complexes containing a few million atoms with shadows, ambient occlusion. Even the depth-of-field focal blur effect can be rendered in real-time on recent hardware. The interactive ray tracing engines use progressive refinement to allow users to manipulate the view using the mouse, and once the mouse is released, the image is refined with monte carlo sampling of lighting, focal blur, and so on. The interactive ray tracing engines allow multiple views to be saved at any time, as well as the default behavior of saving the final image shown when the user exits the viewer. Future versions of VMD will more closely integrate interactive ray tracing, enabling interactive ray tracing in the main VMD window, as an alternative to the traditional OpenGL-based visualization engine in VMD.
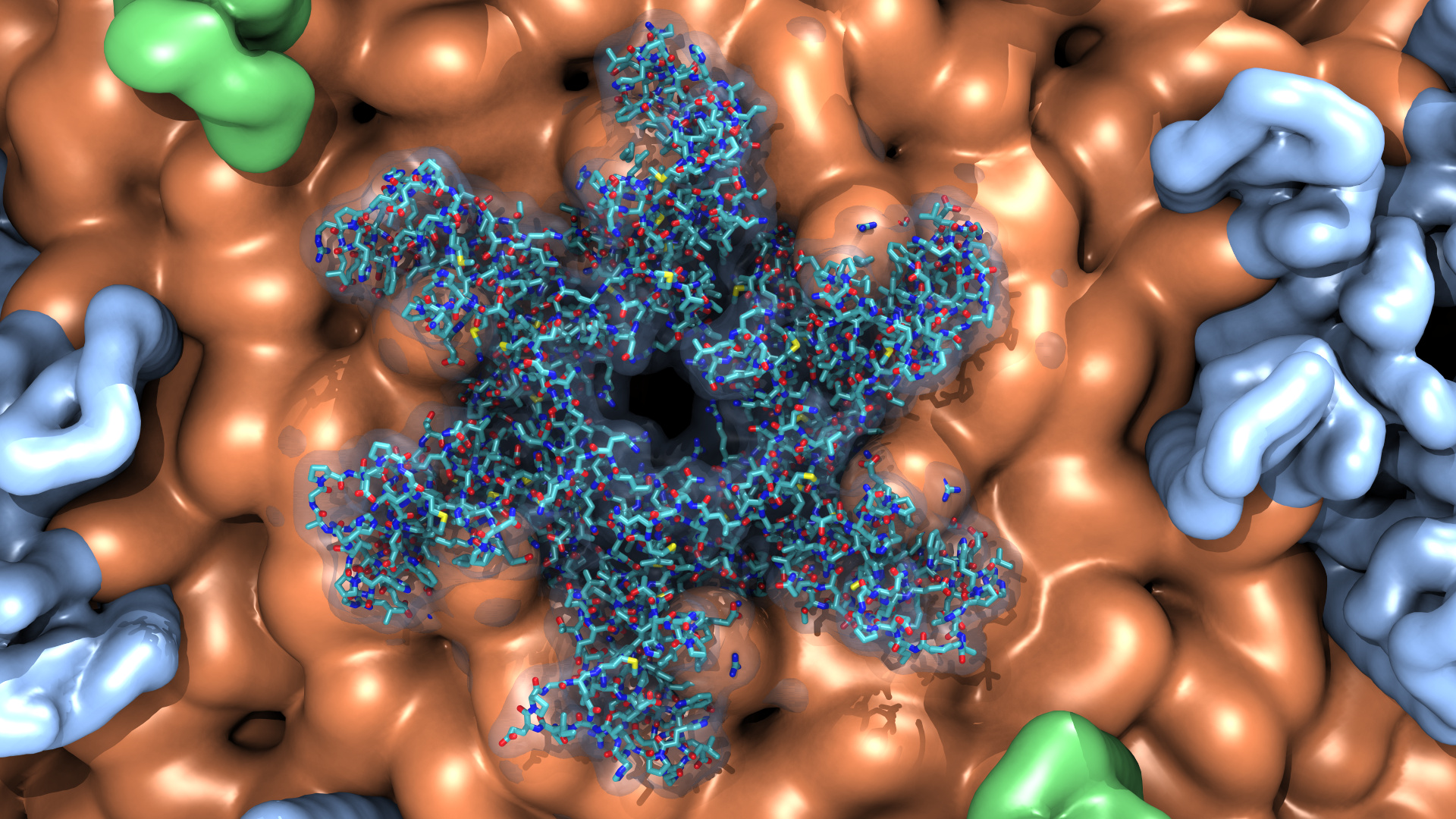
As reported in several publications, VMD uses GPUs to accelerate many of the most computationally demanding visualization and analysis features of interest to biomedical researchers. VMD 1.9.3 advances these capabilities further, adding support for the latest generation of NVIDIA "Pascal" GPUs. The new version includes an improved GPU-accelerated implementation of the QuickSurf molecular surface representation which can now handle rendering of large biomolecular complexes and cellular organelles containing several hundred million particles, with significant improvements for the display of coarse-grained models of such as proto-cell membranes. The QuickSurf representation enables smooth interactive animation of moderate sized biomolecular complexes consisting of a few hundred thousand to one million atoms, and interactive display of molecular surfaces for multi-million atom complexes such as virus capsids. The range of acceleration provided by GPUs depends on the capabilities of the specific GPU device(s) installed, and the details of the calculation. Typical (approximate) acceleration factors for the algorithms in VMD comparing a single high-end GPU vs. a single quad-core CPU are: molecular surface display 2.5x to 11x, electrostatics 5x to 11x, implicit ligand sampling 4x to 7.5x, calculation of radial distribution functions 7.5x to 22x, molecular orbital calculation 10x to 30x. Details on making best use of the GPU acceleration capabilities in VMD are provided here.
Fast "QuickSurf" multi-resolution molecular surface
calculation and display with CUDA GPU accelerationVMD 1.9.3 adds support for the new Cray XC50 supercomputer with compute nodes that combine the latest Intel Xeon CPUs and NVIDIA P100 GPU accelerators utilizing die-stacked memory. The new version of VMD also adds support for workstations, clusters, and supercomputers based on the new IBM POWER8 (OpenPOWER) and Intel Xeon Phi (Knight's Landing) CPUs to be used in the DOE/ORNL Crest (OpenPOWER/NVLink/Tesla P100), NSF/TACC Stampede-2 (Dell/Intel KNL), and DOE/ALCF Theta (Cray/Intel KNL) systems. New hand-vectorized CPU kernels for commonly used VMD analysis and visualization algorithms increase performance significantly beyond standard C++ on both platforms, making advantageous use of the native VSX and AVX-512 vector instruction sets on each processor, respectively.

VMD 1.9.3 supports the new CPUs and GPUs used in the latest
clusters and supercomputers at NSF/TACC Stampede-2 (left),
DOE/ALCF Theta (right), DOE/ORNL Crest, NSF/PSC Bridges-2,
and the new CSCS/ETHZ Piz Daint Cray XC50.The new OpenPOWER version of VMD supports CUDA GPU acceleration for key analysis kernels, and benefits from the large memory capacity and memory bandwidth available on these systems, as reported here. VMD takes advantage of the high performance NVLink CPU-GPU interconnect on the new IBM "Minsky" platform containing POWER8 CPUs paired with NVIDIA Tesla P100 GPUs, shown with some early performance results here
The new Intel Xeon Phi AVX-512 version of VMD has been developed to exploit the wide 16-element SIMD units for key analysis and visualization tasks, incorporating new hand-written AVX-512 analysis kernels and support for the Intel OSPRay ray tracing engine optimized for the new Xeon Phi processors, as shown with some early performance results here.
VMD 1.9.3 incorporates initial support for a variety of ARM processors paired with GPU accelerators (on-chip and/or add-in-board). Due to lack of standardization among ARM operating system distributions, ARM platforms currently require compilation from the VMD source code. Results of early performance tests on GPU-accelerated ARM platforms and associated optimizations have been reported here.
One of the features of VMD 1.9.3 is support for off-screen rasterization on clouds, clusters, and supercomputers, through the use of the latest Embedded GL (EGL) graphics APIs for managing OpenGL contexts and rendering surfaces. With EGL, it is no longer necessary for a compute node to have a complete windowing system installation to be able to perform routine post-hoc molecular visualization tasks in place on compute nodes where the data are generated and stored, thereby eliminating the need for time consuming data staging and file transfer activities. The use of EGL eliminates the need for special compilations of VMD for GPU-accelerated or software-based rasterization, eliminating many of the technical hurdles and administrative tasks that would have previously been required for deployment of a full OpenGL-enabled VMD installation on large parallel computers. The new EGL-based graphics feature in VMD supports all of the standard VMD OpenGL features including pixel-rate lighting, multisample antialiasing, high quality transparency, text rendering, and all of the standard VMD materials and shading features, as reported here.
VMD 1.9.3 adds support for off-screen rasterization on clouds, clusters,
and supercomputers without the need for installing or running
windowing system software using the Embedded GL (EGL) graphics API.