How to run IMD
How to run Interactive Molecular Dynamics
Step-by-step instructions
-
Obtain a copy of VMD ,
version 1.4b1 or higher, and a copy of
NAMD2 , version 2.1b1
or higher.
-
Append the following lines to your NAMD2 startup file:
IMDon yes IMDport 2030 IMDfreq 1
Assuming your system starts up ok, NAMD will wait for VMD to connect before proceeding with the simulation. IMDport tells NAMD which port to listen on for VMD's connection. IMDfreq tells NAMD to pass coordinates to VMD at every timestep.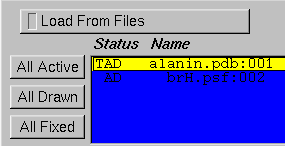
-
Start VMD and load the same system you're simulating in NAMD.
You can load the molecule in any format VMD supports, but there must be
as many atoms in the structure file you load as there are in the .psf file
used by NAMD, and the atoms must be indexed in the same way. The safest
way to ensure that this is the case is to use the same .pdb, .psf, or both
as was used by NAMD.
VMD assumes that the top molecule is the one which is running on the remote machine. Whatever molecule you last loaded should already be top. You can set the top molecule using the Molecule Form, a portion of which is shown above; in this figure, alanin.pdb is the top molecule because of the T to the left of its name.
-
Assume you are running NAMD on the machine named titan , and the
IMDport in the NAMD startup file is 2030. Connect to the simulation by typing
the following from the VMD command line:
vmd> imd connect titan 2030
If you are running NAMD on multiple nodes (such as on a Beowulf cluster), you must enter the machine corresponding to node 0 in the simulation. This can be determined by looking at the second line of the output file from NAMD.Other commands to control the connection are:
- imd detach : Detaches from the simulation, leaving it to run.
- imd kill : Detaches from the simulation and tries to terminate it.
- imd keep rate : Sets the rate at which VMD saves simulation frames to the given rate; these frames can be played back at any time using the Animate form, or saved to a DCD file.
- imd transfer rate : Sets the rate at which NAMD sends coordinates to VMD.
- If the simulation crashes or otherwise needs to be restarted, try simply restarting NAMD the same way it was started before. You may also have to detach VMD before you can reconnect to the simulation. If you have saved frames from previous simulations, these will be preserved.
Tk widget for IMD
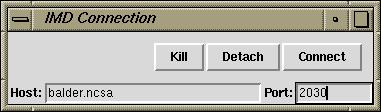 This Tk widget may make more convenient to
connect and disconnect to remote systems. Download the script and
source it from within VMD. Pressing enter after from within
any of the text fields will cause VMD to try to connect to the simulation.
This Tk widget may make more convenient to
connect and disconnect to remote systems. Download the script and
source it from within VMD. Pressing enter after from within
any of the text fields will cause VMD to try to connect to the simulation.
Example scripts
Below are files you can download to test out IMD on your system.alanin.pdb The pdb file.
alanin.psf The psf file.
alanin.conf The NAMD startup file.
alanin.params The Charmm parameter file used by NAMD.
Download all these files into a single directory. Start NAMD using the alanin.conf file, then load a molecule using the alanin.psf and alanin.pdb files. Follow the above instructions to connect to the simulation, and watch and watch the action! When you're ready for more fun, you can apply forces using the mouse, or with a 3D tracker.


