Algorithms
Molecular Dynamics
Atomic positions obey Newton's second law
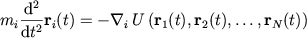
|
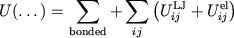
|
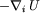 is very time consuming.
To speed up these calculations,
NAMD uses:
is very time consuming.
To speed up these calculations,
NAMD uses:
- parallelism,
- a fast multipole algorithm, and
- multiple time stepping (MTS).
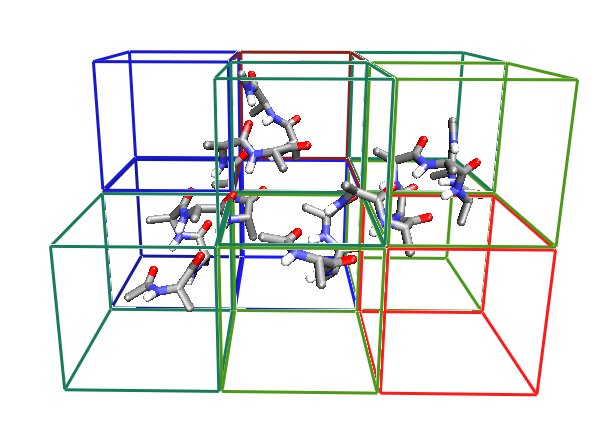
Spatial Decomposition
NAMD divides the simulation space into rectangular regions called patches. Each patch is responsible for updating the coordinates of the atoms contained in its region of space. Patch dimensions are chosen to be greater than the cutoff radius for non-bonded interactions, which eliminates the need for communication between non-adjacent patches. A cluster of adjacent patches is assigned to each processor. This design reduces the amount of interprocessor communication, enabling scalable parallelism.
| particles |
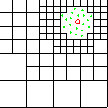
|
level 3 |
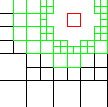
|
|---|---|---|---|
| level 4 |
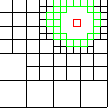
|
level 2 |
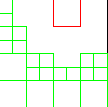
|
Enhanced Parallel Fast Multipole Algorithm
(Board et al. 1995)
The effect of a charged particle is represented directly at a short length scale. At longer length scales its effect is pooled with other charges and represented by a multipole expansion, which is transfered to distant cubes and expressed as Taylor expansions.
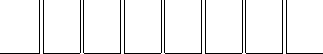
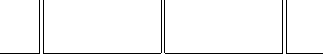
Verlet-I / r-RESPA / Impulse MTS Method
Forces are approximated by a sequence of impulses:
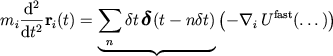
|
|
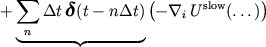
|
|
Partitioning the potential energy as
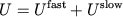
|
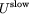 consists of long-range electrostatics,
consists of long-range electrostatics,
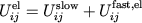
|
|
|
|---|---|
| slow part | fast part |
Mollified Impulse Method
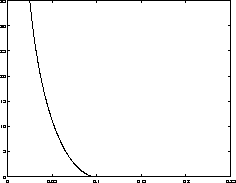
The Impluse MTS method is unstable unless:
slow-force
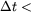 half the period of the highest frequency fast-force motion
half the period of the highest frequency fast-force motion
|
This is overcome by the Mollified Impulse method (MOLLY).
Define vibration-averaged positions
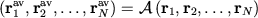
|
 by
by
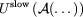 in the Impulse MTS method.
in the Impulse MTS method.
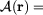 projection in configuration space of positions
onto equilibrium manifold for
projection in configuration space of positions
onto equilibrium manifold for
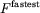
|
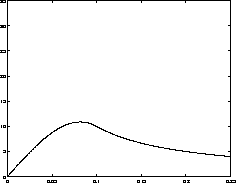
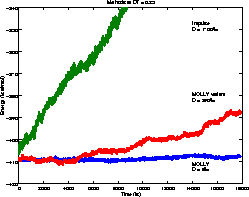
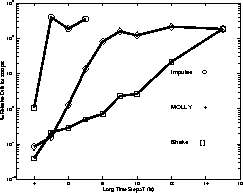
Some graphical MOLLY results:
|
|
|---|---|
total energy vs. time for
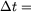 6.25 fs
6.25 fs
|
percent relative drift vs. time |
Timesteps for Different Interactions
For a harmonic oscillator:
| choosing |
1/12
|
| gives |
frequency error
 1.14%
1.14%
|
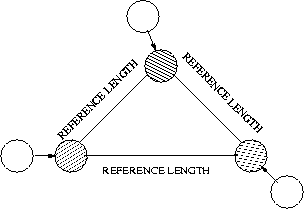
The generalized period for an interaction is
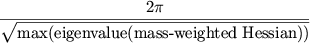
|
|---|
Examples of
 :
:
|
Normal mode analysis of 125 water molecules |
period(s) |
|
|---|---|---|
| bond length stretching | 9.9, 10.1 fs | 0.8 fs |
| bond angle bending | 18.9 fs | 1.6 fs |
|
Simulations measuring the generalized period of the Lennard-Jones potential for 125 water molecules at 300 K |
total time |
worst case
|
|---|---|---|
| 10 fs | 2.3 fs | |
| 100 fs | 2.0 fs | |
| 1000 fs | 1.7 fs |
Avoiding Distance Calculations
By construction
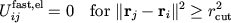
|
|---|
To avoid most redundant distance calculations,
monitor atom movement after patch assignment.
Choose
 to be the maximum distance traveled by an atom.
to be the maximum distance traveled by an atom.
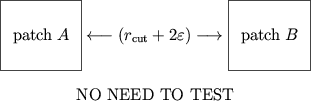
|
|---|
Multiple Grid N-body Solvers
These are interpolation-based solvers which utilize a splitting of the potential into short-range and smooth long-range parts:
- short-range contribution: direct calculation using spatial hashing
- long-range contribution: approximate fast matrix-vector product on a grid using a multilevel method
The motivation is to be faster than the fast multipole algorithm:
- relative simplicity of the algorithm enables the avoidance of black box overhead
- continuity of forces results in stable time stepping
- continuity of force decomposition results in stable multiple time stepping
The following tables compare an initial implementation of
the multiple grid N-body solver with DPMTA (Board et al.),
a fast multipole implementation.
The test system was a 60 Angstrom box of water containing
a total of 20544 atoms.
The timings were performed after compiling both codes with the
cc -fast option on a Sun Ultra-60.
| Multiple grid N-body solver (cutoff radius = 8 Angstroms) | ||||
|
grid spacing (Angstroms) |
average force error (%) |
maximum force error (%) |
energy error (%) |
time (seconds) |
|---|---|---|---|---|
| 4.36 | 0.29 | 1.32 | 0.0013 | 1.87 |
| 2.77 | 0.17 | 0.67 | 0.0024 | 2.95 |
| 2.03 | 0.14 | 0.58 | 0.0017 | 8.15 |
| DPMTA, fast multipole implementation (theta = 0.75) | ||||
|
number of terms |
average force error (%) |
maximum force error (%) |
energy error (%) |
time (seconds) |
|---|---|---|---|---|
| 4 | 0.30 | 1.83 | 0.0004 | 6.49 |
| 8 | 0.02 | 0.25 | 0.0002 | 8.42 |
Fast Matrix-Vector Product on a Grid
The electrostatic potential energy due to N charged particles is
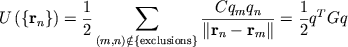
|
|---|
To compute the long-range contribution, we need to form the product
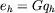 which is obtained by approximating the smooth part of
G on a grid of spacing h.
which is obtained by approximating the smooth part of
G on a grid of spacing h.
A fast way to do this is to approximate the smooth part of
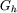 on a coarser grid with spacing 2h.
Using superscript k to represent a grid with spacing
on a coarser grid with spacing 2h.
Using superscript k to represent a grid with spacing
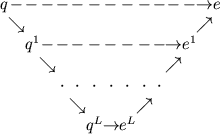
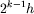 the computation through L grid levels is represented schematically:
the computation through L grid levels is represented schematically:
|
|---|


