Sepehr Dehghani-Ghahnaviyeh
I am a Graduate Research Assistant at the Theoretical and Computational Biophysics Group (TCBG) of the Beckman Institute at the University of Illinois at Urbana-Champaign. As a computational biophysicist, I have been involved in multiple biological and software development related projects. Particularly, I am interested in large scale conformational changes of proteins. I am working on two different proteins at the moment, P-Glycoprotein (a protein known to cause MDR in cancer cells) and Prestin (a membrane protein in hearing system with electromechanical functionalities). Such conformational changes are very important in drug discovery/design fields, in order to target these two proteins. Furthermore, I am extremely interested in software development in different computational and molecular biology areas, particularly I have been developing a machine learning toolkit to study protein oligomerization using Variational Autoencoder (VAE) models. Also, I am a member of NAMD developers team, leading the efforts to implement different features in both NAMD and VMD to simulate Coarse Grained Molecular Dynamics systems. I have been also part of an effort to develop an integrative toolkit, known as MMP (Membrane Mixer Plugin) which is already implemented in VMD, to shuffle lipids in heterogeneous membranes and generate multiple healthy and ready-to-use replicas of different membrane/membrane-protein systems.
Home Department: Computational Biophysics and Quantitavie Biollogy.
Office Address: Beckman Institute, Room 3117.
Office Phone: (217) 244-1851
Email Address: sepehrd2@illinois.edu
Follow @dehghani_sepehrProfessional Experience
- Co-op at the computational scinece team of Biogen - Biogen - MA, USA
- Co-op at the computational scinece team of Moderna - Moderna - MA, USA
I was a co-op at the computational science team of Biogen (January 2022 - May 2022). My project involved development of a platform to automate Free Energy Perturbation (FEP) in AMBER. This platform will be further used for calculating binding affinity of different small molecules to RNA.
I was a co-op at the computational science team of Moderna (January 2021 - June 2021). I was involved in multiple projects, the most important of which was simulating Lipid Nanoparticles (LNPs) containing novel ionizable lipids, employing Molecular Dynamics (MD) simulations. The LNPs are mainly used in mRNA therapeutics and my investigations were part of the LNPs assembly project of Moderna. Furthermore, I was investigating the effect of different novel lipids in mRNA encapsulation, employing MD, enhanced sampling, and data science related techniques.
Education
- PhD in Computational Biophysics, Minor in Computational Science and Engineering - University of Illinois at Urbana-Champaign - IL, USA
- Guest M.Sc. Student - Inistitue of Complex Systems and Advanced Simulation - Forschungszentrum Juelich, Germany
- M.Sc. in Mechanical Engineering - Sharif University of Technology - Tehran, Iran
- B.Sc. in Mechanical Engineering - Iran University of Science and Technology - Tehran, Iran
Advisor: Prof. Emad Tajkhorshid
Advisor: Prof. Gerhard Gompper and Dr. Thorsten Auth

Selected Publications
1.Hanbin Jeong, Sarah Clark, April Goehring, Sepehr Dehghani-Ghahnaviyeh, Ali Rasouli, Emad Tajkhorshid, and Eric Gouaux. Structure of C. elegans TMC-1 complex illuminates auditory mechanosensory transduction; Nature, In Press (2022).
2. Reza Dastvan, Ali Rasouli*, Sepehr Dehghani-Ghahnaviyeh*, Samantha Gies, and Emad Tajkhorshid. Protonation-dependent alternating access of a spinster transporter, an emerging family of broad-specificity efflux pumps; Nature Communication, In Press (2022).
2. Giuseppe Licari*, Sepehr Dehghani-Ghahnaviyeh*, and Emad Tajkhorshid; Membrane Mixer: an integrative toolkit to shuffle lipids in heterogeneous membranes; Journal of Chemical Information and Modeling 62.4 (2022): 986-996.
3. Jingpeng Ge, Johannes Elferich, Sepehr Dehghani-Ghahnaviyeh, Zhiyu Zhao, Emad Tajkhorshid, and Eric Gouaux; Molecular mechanism of prestin electromotive signal amplification; Cell 184.18 (2021): 4669-4679.
4. Sepehr Dehghani-Ghahnaviyeh, Karan Kapoor, and Emad Tajkhorshid; Conformational changes of the nucleotide binding domains of P-glycoprotein induced by ATP hydrolysis; FEBS letters 595.6 (2021): 735-749.
5. Sepehr Dehghani-Ghahnaviyeh, Zhiyu Zhao, and Emad Tajkhorshid; Lipid-mediated organization of prestin in the cochlear membrane and its implications in sound amplification; bioRxiv (2022).
7. Le Thi My Le, James R. Thompson, Sepehr Dehghani-Ghahnaviyeh, Shashank Pant, Phuoc X. Dang, Takahisa Kanikeyo, Emad Tajkhorshid, Amer Alam. Capturing the interplay of membrane lipids and structural transitions in human ABCA7; bioRxiv (2022).
8. Ali Rasouli*, Qin Yu*, Sepehr Dehghani-Ghahnaviyeh, Po-Chao Wen, Julia Kowal, Kaspar Locher, and Emad Tajkhorshid. Differential effects of cholesterol and phospholipids on ligand binding of multidrug transporter ABCG2; Under Reveiew (2022).
9. Karan Kapoor, Sepehr Dehghani-Ghahnaviyeh, and Emad Tajkhorshid; Local Switches Regulating the Global Conformational Transitions in P-Glycoprotein; In Prepreation
10. Clara Abaurrea Velasco*, Sepehr Dehghani-Ghahnaviyeh*, Hossein Nejat Pishkenari, Thorsten Auth, and Gerhard Gompper; Complex self-propelled rings: a minimal model for cell motility; Soft Matter 13, no. 35 (2017): 5865-5876
* equal contribution
Research Interests
-
Large Scale Conformational Changes in Proteins
-
Using Molecular Dynamcis and Monte-Carlo Techniques to Develpe a Toolkit in VMD to Generate Ready-to-use Atomic Systems
-
Memrane Fusion in Synapses
-
Developing a Machine Learning Toolkit to Study Protien Oligomerization
-
Coarse Grained Molecular Dynamics in NAMD
Research Highlights: P-glycoprotein
In this project, P-glycoprotein local, as well as global conformational changes due to ATP hydrolysis are investigated, using unbiased MD simulations of Pgp in multiple nucleotide-bound states. A total of five different systems, including all possible combinations of ATP and ADP bound to the NBDs along with the apo system (nucleotide-free) are constructed in their membrane-bound forms and simulated. Important nucleotide binding domains (NBD) functional motifs, triggering the ATP hydrolysis are explored and analyzed. Our results suggest that local conformational changes in the NBDs take place mostly within the signature motif followed by abolishing of nucleotide (ATP and ADP) interactions with different motifs and loops at the nucleotide binding site, whereas global conformational changes trigger NBDs dissociation through changes in their relative orientation. Additionally, our simulations show that the E-loop can be considered as the catalytic base in Pgp, trapping a catalytic water and generating a hydrolysis-competent state optimal for ATP hydrolysis. Through the course of the simulations, multiple hydrolysis-competent states were captured with the catalytic water present in either NBD, suggesting that in Pgp both NBDs are capable of ATP binding and hydrolysis. Our result suggest that the global and local conformational changes in NBDs, as well as NBD dissociation may be initiated from the captured catalytic states and as a result of ATP hydrolysis, and nucleotide dissociation.
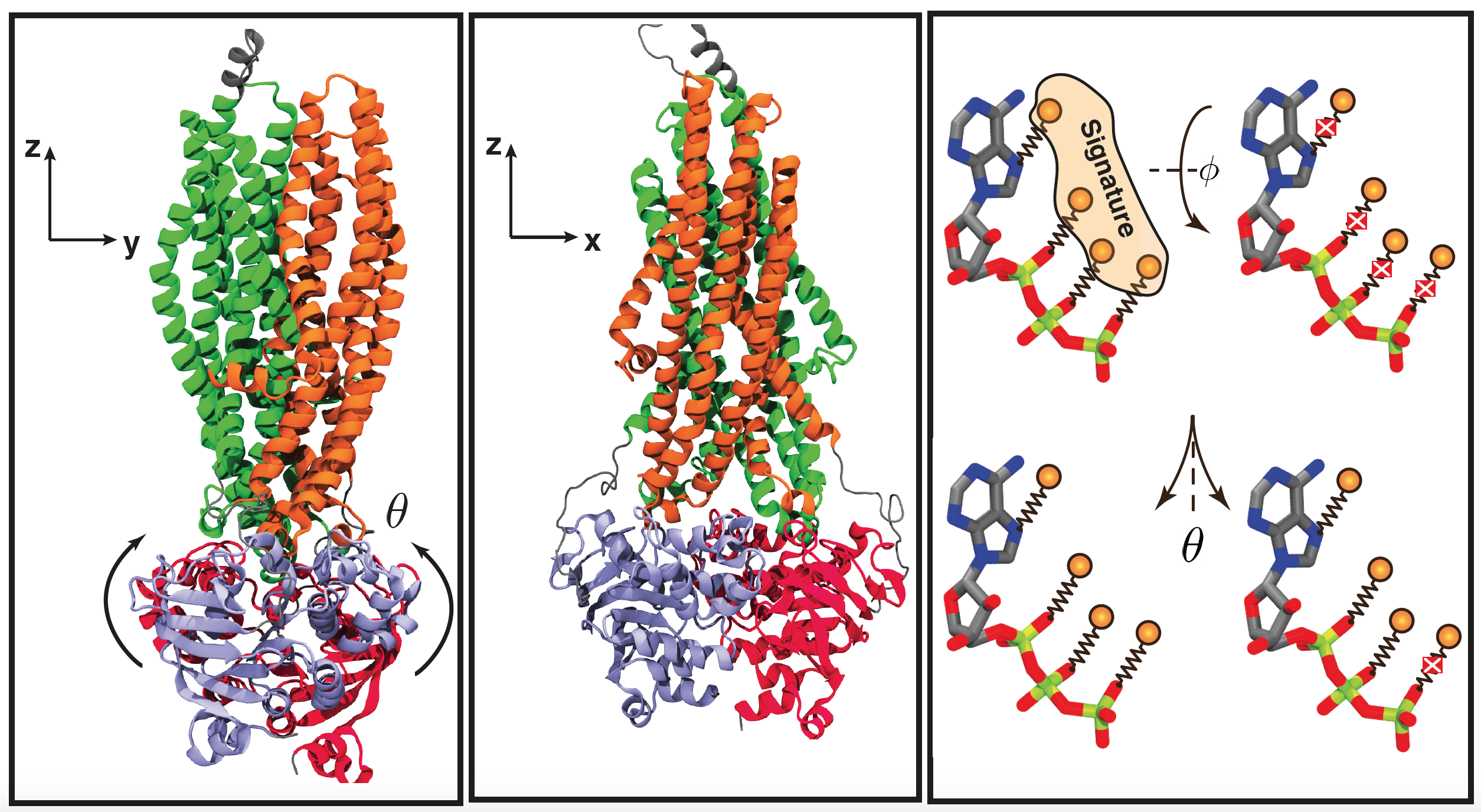
Research Highlights: Membrane Mixer Plugin (MMP)
In this project, we are introducing a method that has been developed in our lab to rapidly generate new membranes with shuffled lipids and to provide multiple initial configurations within minutes. As there are already many tools that can build complex membranes, our aim was to conceive an algorithm that would use an initial membrane to generate several equilibrated and ready-to-use replicas with different lipid positioning in a short time maintaining the initial topology and concentration of lipids. The fully-automated algorithm developed in our work is similar to some extend to the replacement method, but fundamentally differ from it in the application of an external potential that reduces clashes between atoms before shuffling the lipids. This method allows for any lipid exchange in the membrane, irrespective of their molecular weight and type and includes four steps: (1) randomly selecting some of the lipid molecules based on the exchange rate requested by the user, (2) applying grid-based potentials on chosen lipids to make some space before the exchange process, (3) exchanging lipids and removing the grid potentials gradually through several equilibration runs, (4) check and fix possible ring piercing. %In fact, the external potential acts by generating the required space %for the exchange to occur with reduced energy penalties. The algorithm has been tested on several applications that encompass most used cases in the MD community. Analyses performed on the shuffled membranes show that the method can reliably generate bilayers with virtually indistinguishable physico-chemical properties from the initial membrane. This method has been implemented in the latest version of VMD as "Membrane Mixer 1.0'' plugin (MMP). Details about the plugin can be found here.

Research Highlights: Membrane Fusion in Synapses
This is an going project with the goal of characheturing the biomechanics of SNARE mediated vesicle fusion at an atomic level. The project leverages computational methods to combine structural data from multiple experimental sources of differing resolutions (X-ray crystallography of individual pro- teins, medium-resolution cryo-EM of multi-protein systems, and low-resolution cryo-EM tomography of subcellular organelles) yielding atomic-resolution structural and dynamical description of the SNARE-mediated vesicle fusion. We have designed our own in-house scripts to model Vesical and Plasma membranes with different sizes and lipid compositions. The stability of the assembled multicomponent system is already tested through short Molecular Dynamics simulation. To the best of our knowledge, this system is the first stable atomistic model of the SNARE complex in the pre-fusion state. Furthermore, we have employed a non-equilibrium Grid Potential protocol to facilate the membrane fusion process, where two independent grids will be used to induce the changes in the membrane separation and SNARE complex, respectively, in a linear manner. The grid geometry also is changed during the non-equilibrium simulation in order to keep the system as stable as possible. The entire process can allow us to generate intermediate states (seeds) between the pre-fusion and post-fusion states.

Research Highlights: A Machine Learning Toolkit to Study Protein Oligomirization
In this project, I have been working on a python code capable of constructing VAE models, in order to study protein-protein interactions and oligomerization. The code inputs a multidimensional matrix, containing the pairwise distances between amino acids of different monomers, which need to be calculated by users from a molecular dynamics (MD) simulation. The package provides a parallel framework for the users to perform this distance calculation, which can be run on multiple processors. The implemented machine learning module then uses this input matrix to construct a VAE model to map the data into a latent space. Different hyperparameters of the module including, the deep neural network layers and nodes in the VAE model, learning rate, number of epochs, and the dimension of the latent space can be optimized, using Hyperopt libraries in python. The VAE model itself takes advantage of different Pytorch libraries, including: nn and autograd, and can be run on GPU/CPU. The latent space constructed here can contain several clusters, representing different protein oligomerization states, as well as their differences and similarities with amino acid resolution. It is important to mention that the oligomerization states each should be sampled in the MD simulation, otherwise, the implemented VAE algorithm cannot predict them. We have tested this module on a 40 microseconds KRAS dimer trajectory (simulated on Anton2 machine), and the constructed latent space was able to show all the dimerization states of KRAS monomers and their similarities and differences (unpublished - in preparation). The main code, as well as the related files can be found here.

Research Highlights: Coarse Grained Molecular Dynamics in NAMD
This project is about an ongoing effore to implement necessary features in NAMD to simulate coarse grained molecular dyanmics system based on MARTINI forcefiled. I have been working tightly with the NAMD developers team to implement necessary routines in the NAMD source code to define different form of constraints in the Coarse Grained structures, as well as generate virtual sites, introduced in MARTINI 2.2 forcefiled. The project have many challenges that need to be addressed. At the moment, the biggest challenge is to rearrange the data structure of the coarse grained beads x,y, and z positions in a way to inlcude the beads belonging to a constrint or virtual site to one computing batch to fix the communication problem between computing units. (unpublished - in preparation).

Programming
I have been working on several machine learning, CUDA programming, Molecular Dynamcis, and Monte-Carlo codes and packages. All the codes can be found here in my Github profile. Some of the codes are listed bellow: