Molecular Replacement
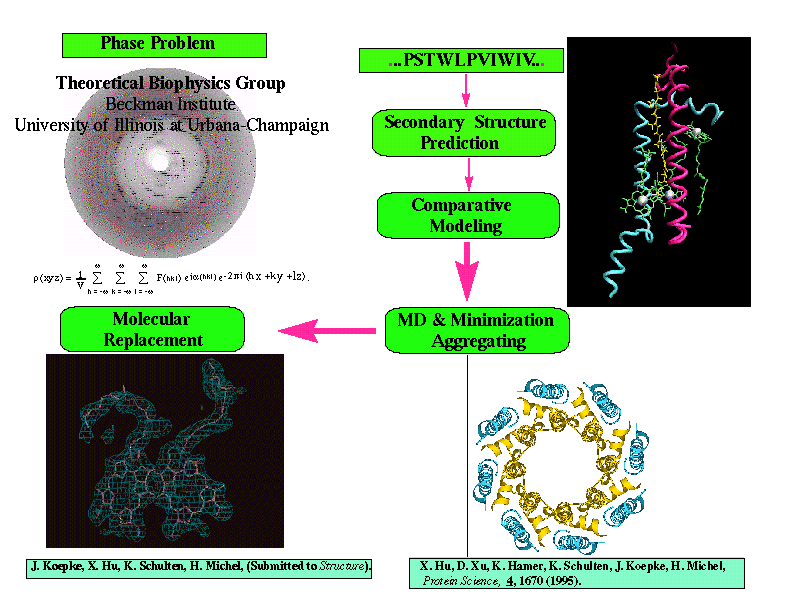
Overview of ab initio molecular replacement method as applied to structure determination of LH-II of Rs. molischianum.
We are aiming at developing the method of ab initio molecular replacement for x-ray crystallographic structure determination of proteins. The ab initio molecular replacement method involves modeling of tertiary structures for a protein and utilization of the predicted structures as a probe model in the framework of the so-called molecular replacement method [1,2]. Prediction of the initial protein structure and subsequent refinements entail the use of well-known secondary structure prediction techniques (to find basic folding patterns from the protein amino-acid sequence), and methods for determining the tertiary folding pattern such as homology searches, threading, and potential of mean force (PMF) calculations. Starting from these data, MD refinement can be used to fill in the positions of amino acid side chains, and to equilibrate and minimize the structure.
More than 50,000 sequences for proteins are known up to date (SWISSPROT release 33.0), but only about 4,300 protein structures have been solved (PDB Release 77) and these structures represent roughly 100 different folding topologies (CATH database). Coding sequences can now be determined relatively easily by molecular genetic methods. However, determination of three dimensional structures, mainly by x-ray crystallography, is a much more complex and slower task [3]. One can clearly discern a rapid widening of the gap between sequence data gathered and their conversion into atomic coordinates of the underlying structures.
Computational methods have already played an enormously important role in speeding up the conversion, but a key bottleneck remains the so-called phase problem in x-ray crystallographical structure determination [3]. To resolve a structure from measured diffraction intensities requires knowledge of phases which is unobtainable from a single diffraction experiment. Conventionally, the phase problem is solved by means of the multiple isomorphous replacement method [4] which requires two or more isomorphous heavy metal derivatives. An alternative solution to the phase problem is to phase the structure by using a homologous structure in a procedure called molecular replacement [1,2]. In this method, a homologous probe structure is fit into the unit cell of the unknown structure and used to generate an initial phasing model for the unknown structure. Typically, a homology search of the PDB database is performed to seek an existing protein structure as the probe structure.
Crystallization of isomorphous heavy metal derivatives is a tedious and sometimes impossible task. The conventional molecular replacement method relies heavily on the availability of the structure of a highly homologous protein. It is not uncommon that both approaches fail. To extend the applicability of the x-ray diffraction method, we propose to develop the method of ab initio molecular replacement. Unlike the conventional molecular replacement method, the probe model is modeled utilizing protein structure prediction methods rather than the structure of a homologous protein.
If successful, the proposed method will broaden the class of solvable protein structures and speed up the process of x-ray crystallographic structure determination.
We have recently determined the structure of the Light-Harvesting Complex II (LH-II) of Rhodospirillum molischianum by means of molecular replacement utilizing a computationally modeled structure as a probe model [5,6,7]. It is a significant step towards the ab initio molecular replacement method.


