Protein Domain Movement
We have developed a new algorithm, Hingefind, for the identification of domain movements in proteins. The method partitions a protein in domains of preserved packing by comparing two structures. The well-fitting substructures are extracted by a "adaptive selection" procedure using least-squares fitting. The procedure allows to maintain the connectivity of the substructure and to filter significant structural differences (movements of rigid domains) from experimental noise in the compared structures of the rigid domains. The algorithm then characterizes the domain movements between the found rigid domains by effective hinge-axes. Currently, the technique is implemented in the script languages X-PLOR (Axel T. Brunger, 1992) and Tcl (John Ousterhout, 1988). The output files of the X-PLOR version can be visualized with standard graphics packages, e.g. VMD or Quanta. The Tcl version is designed for interactive use with the program VMD. We have applied the method to several known instances of domain movements in protein structures from computer simulations or for which there is crystallographic evidence for the movement. We have compared structures of actin, hexokinase, lactoferrin, and the extracellular domains of human tissue factor and of the receptor of human growth factor. The compared structures exhibit a variety of hinge and shear motions depicted by the algorithm. The method should be a useful tool to analyze domain movements in crystallographic structures or structures from computer simulations.
Characterization of Domain Motions
Proteins are empowered by their intrinsic flexibility to a wide spectrum of biochemical function in catalysis, regulation, protein assembly and cell motility. Physiological agents control the spatial arrangement of moving domains of preserved topology in different protein conformations. In a recent survey, Gerstein et al. investigated domain movements of all known cases with existing crystallographic evidence (Gerstein et al., Biochemistry 33 (1994), 6739-6749). Their survey is documented in the Protein Motions Database at Yale. Of special interest are hinge-bending movements, where rigid domains are connected by flexible joints which tether the domains and constrain their movement. Hinge-bending is believed to allow an induced fit of molecular surfaces in protein assembly and ligand docking. In a conformational analysis of protein structures the identification of hinge axes and their corresponding rotation angles permits a useful representation of protein domain movements.
Based on this concept of protein domain movements we have developed Hingefind to investigate such movements by comparing two protein structures. The algorithm allows to partition a protein into rigid domains of preserved packing depending on a prespecified resolution controlled by the user. The conformations being searched for domains must have their sequence aligned by identifying pairs of corresponding residues. No further assertions are made regarding the extent of rigid domains and the movements between them.
Click here to get a 77 kByte image of actin domain movements characterized by Hingefind:
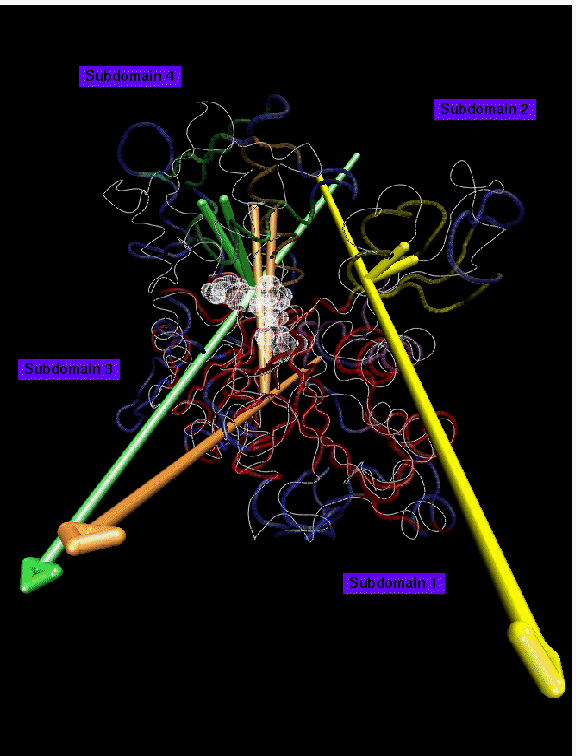
Backbone trace of a actin structure from MD simulation presented in color, crystal structure (PDB entry 1ATN) in white. ADP nucleotide atoms shown as transparent spheres. 5 domains larger than 10 residues have been found at 1.8 A resolution: domain 1 (red, 166 residues) is the reference domain; domain 2 (green, 26 residues) rotates by 14 degrees; domain 3 (yellow, 24 residues) rotates by 21 degrees; domain 4 (purple, 22 residues, hingeaxis not shown); domain 5 (orange, 17 residues) rotates by 5 degrees. Disordered regions are shown in blue. The movement of subdomain 2 in the simulated structure can be explained as a relaxation effect due to the missing DNase1 of the complex in the crystal structure. The observed movement of subdomain 4 may be due to a compaction of the nucleotide binding site. The relative large number of small domains and the relative large fraction of disordered residues on the surface of the protein (120 of 375 residues) indicate that structures from computer simulations are prone to be distorted by transient tertiary structure rearrangements due to limited simulation time.
Click here to get a 83 kByte image of hexokinase domain movements characterized by Hingefind:
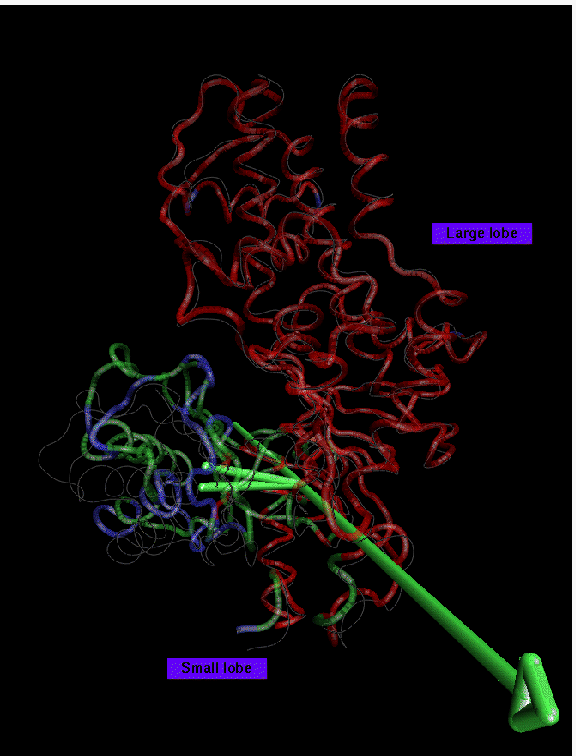
Backbone trace of glucose bound yeast hexokinase (PDB entry 1HKG) presented in color, unliganded structure (PDB entry 2YHX) in white. Glucose not shown. 2 domains larger than 10 residues have been found at 1.8 A resolution: domain 1 (red, 317 residues) is the reference domain; domain 2 (green, 98 residues) rotates by 13 degrees. Disordered regions (42 residues) are shown in blue. The large lobe of hexokinase appears to be structurally well preserved. Only a part of the small lobe, however, is preserved in the movement and the hinge-axis does not lie in the interface between large and small lobe. The type of tertiary rearrangements in the small lobe are similar to the actin case. This Figure suggests that domain closure in hexokinase is the cumulative result of many small relative movements of secondary structure, rather than a rigid-body motion of the small lobe. Nevertheless, the hinge-axis allows for a coarse grained illustration of the movement of the largest well-fitting subset of residues involved.
Click here to get a 87 kByte image of lactoferrin domain movements characterized by Hingefind:
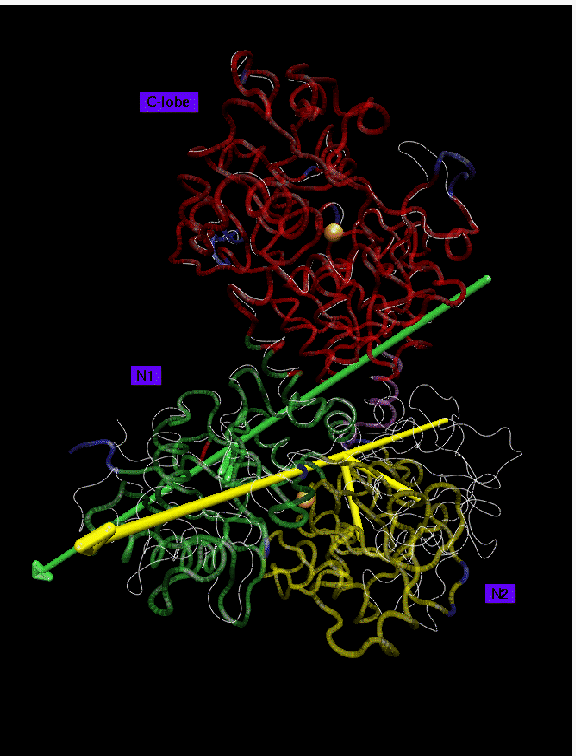
Backbone trace of iron-bound lactferrin (PDB entry 1LFG) presented in color, iron-free form (PDB entry 1LFH) in white. Iron ions are shown in orange. 3 domains larger than 10 residues have been found at 1.2 A resolution: domain 1 (red, 325 residues) is the reference domain; domain 2 (green, 171 residues) rotates by 8 degrees; domain 3 (yellow, 155 residues) rotates by 54 degrees. Disordered regions (26 residues) are shown in blue. The domains in lactoferrin move essentially as rigid bodies. The three found domains are practically identical to the structural domains of lactoferrin, showing a rms deviation of only 0.4 A when superimposed. The axis of rotation of lobe N1 passes through the two beta-strands linking lobes N1 and N2. Lobe N1 rotates only by 8 degrees compared to the 54 degrees of lobe N2. The C-lobe is structurally intact even in the iron free form. The differences in domain movements in the C- and N-lobe have been interpreted as effects of crystal packing forces.
Click here to get a 53 kByte image of cytokinase receptor domain movements characterized by Hingefind:
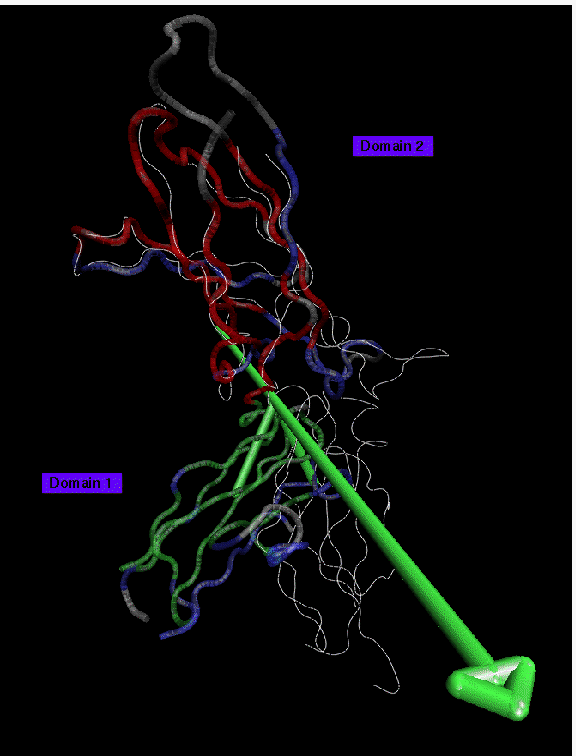
Backbone trace of the receptor of human growth hormone receptor (PDB entry 3HHR) presented in color, crystal structure of human tissue factor (PDB entry 1HFT) in white. 2 domains larger than 10 residues have been found at 3.5 A resolution: domain 1 (red, 54 residues) is the reference domain; domain 2 (green, 53 residues) rotates by 46 degrees. Disordered regions (61 residues) are shown in blue. Regions in the structures where no pair of equivalent residues has been assigned are shown in grey. The case of the two different cytokine receptors demonstrates the challenges of comparing the two homologous proteins of deviating sequence which has been aligned based on J.F. Bazan, PNAS (1990) 87:6934. Even at a low resolution of 3.5 A about one third of the residues appear to be disordered. This result is due to a mismatch in residue pairing from sequence alignment and topological differences in the tertiary structure of the domains. The different orientation of the domains of the two receptors have been attributed to the structure of the interface between the domains: In contrast to the growth hormone receptor the hydrophobic core in tissue factor is continous throughout the interface. As a consequence, the ability of certain residues near the interface to contribute to ligand binding differ significantly between the two receptors.


