TeraGrid '08 Conference Paper
Long Time and Large Size Molecular Dynamics Simulations Made Feasible through New TeraGrid Hardware and Software
Kirby Vandivort, James C. Phillips, Elizabeth Villa, Laxmikant V. Kale, Klaus Schulten, Peter L. Freddolino, James Gumbart, Leonardo G. Trabuco, Danielle E. Chandler, Jen Hsin, Christopher B. Harrison
 This paper was presented at the TeraGrid '08 conference
in Las Vegas and was the winner of the Best Science Track Paper.
This paper was presented at the TeraGrid '08 conference
in Las Vegas and was the winner of the Best Science Track Paper.
You can download the PDF of the paper.
Abstract...
NAMD is a highly parallel, publicly available molecular dynamics program, which has been developed over the past two decades specifically to perform simulations of large biomolecular systems on a broad range of platforms, from laptops to workstation clusters to massively parallel supercomputers.
Advances in NAMD development have been exploited year after year for discoveries in molecular cell biology. Recently, NAMD running on PSC's Bigben and on NCSA's Abe explained the mechanical properties of the blood clotting factor fibrinogen in two 1-million atom simulations. On the water-solvated WW domain, a small 30,000 atom model system also probed in protein folding experiments, NAMD has achieved 101 ns/day on NCSA's Abe, permitting the longest all-atom molecular dynamics simulation ever, stretching over 10 microseconds.
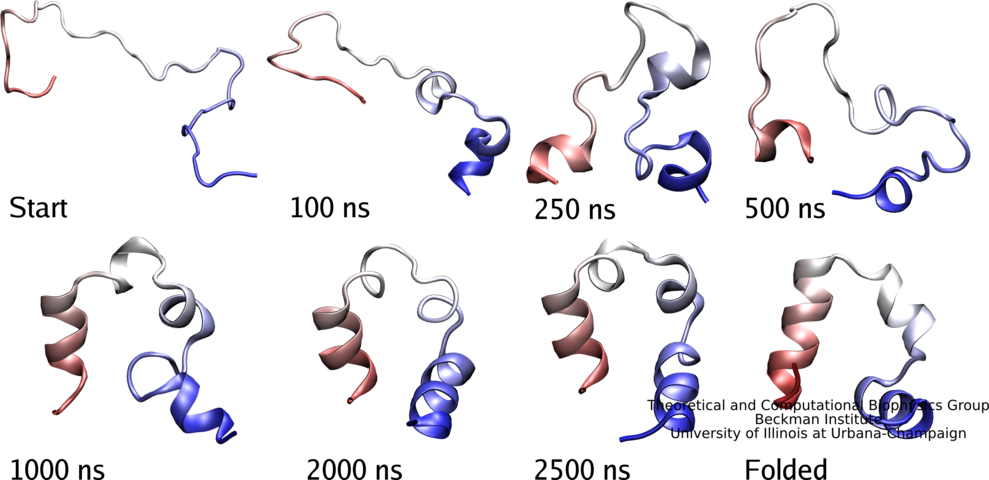 The large size and long time simulations possible now with NAMD,
tuned to the latest teragrid machines Abe NCSA and Ranger TACC,
open up new opportunities in molecular cell biology. This lecture
will report on the new generation of biomedical studies being
accomplished. The Teragrid machines employed for these studies
scale well for small a few thousand atoms as well as for large a
few million atoms systems.
The large size and long time simulations possible now with NAMD,
tuned to the latest teragrid machines Abe NCSA and Ranger TACC,
open up new opportunities in molecular cell biology. This lecture
will report on the new generation of biomedical studies being
accomplished. The Teragrid machines employed for these studies
scale well for small a few thousand atoms as well as for large a
few million atoms systems.
First, folding simulations for three protein systems WW domain, villin head piece, lambda repressor are being completed, sampling in each case three independent trajectories with an expected accumulated time of 50 microseconds.
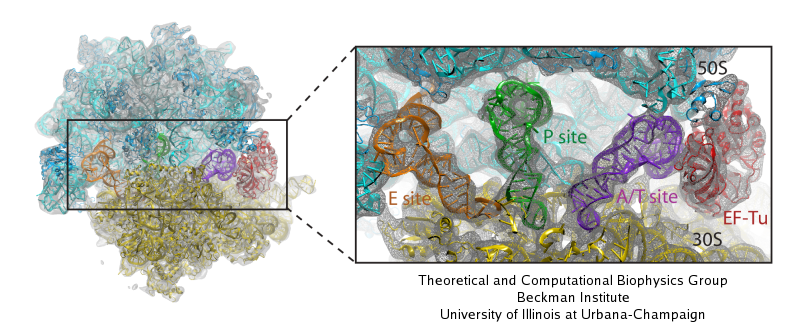 Second, the new hardware and software permit the simulation of
cellular machines composed of many biomolecules. One example is the
ribosome made of RNAs and proteins. Solvated, the ribosome leads to
simulations of three million atoms. Importantly, the all-atom
structures used in these simulations are actually the result of a
massive data analysis that fitted crystallographic structures not
representing functional states of the ribosome into electron
microscopy maps representing functional states. The fitted
structures are then investigated with NAMD in regard to functional
dynamics.
Second, the new hardware and software permit the simulation of
cellular machines composed of many biomolecules. One example is the
ribosome made of RNAs and proteins. Solvated, the ribosome leads to
simulations of three million atoms. Importantly, the all-atom
structures used in these simulations are actually the result of a
massive data analysis that fitted crystallographic structures not
representing functional states of the ribosome into electron
microscopy maps representing functional states. The fitted
structures are then investigated with NAMD in regard to functional
dynamics.
Living systems are hierarchical assemblies of biomolecules; to
understand the very essence of life requires one to understand these
 assemblies in chemical detail, i.e., at the atomic level. The
furthest reach in this regard has been accomplished through a
combination of crystallography, electron microscopy, and atomic
force microscopy that led recently to the atomic level construction
of an entire cellular organelle: the photosynthetic chromatophore in
purple bacteria. These organelles have a diameter of 70 nanometers
and are composed of about 200 proteins that absorb sun light and
store its energy in molecules of Adenosine Triphosphate ATP. A
solvated chromatophore will require simulations of 40 million atoms.
Pieces of the chromatophore involving a million atoms have been
simulated already, revealing a dramatic insight, namely, that the
proteins of the chromatophore bring about the spherical shape of the
organelle and that commonality of curvature controls the protein
assembly as well as organelle size.
assemblies in chemical detail, i.e., at the atomic level. The
furthest reach in this regard has been accomplished through a
combination of crystallography, electron microscopy, and atomic
force microscopy that led recently to the atomic level construction
of an entire cellular organelle: the photosynthetic chromatophore in
purple bacteria. These organelles have a diameter of 70 nanometers
and are composed of about 200 proteins that absorb sun light and
store its energy in molecules of Adenosine Triphosphate ATP. A
solvated chromatophore will require simulations of 40 million atoms.
Pieces of the chromatophore involving a million atoms have been
simulated already, revealing a dramatic insight, namely, that the
proteins of the chromatophore bring about the spherical shape of the
organelle and that commonality of curvature controls the protein
assembly as well as organelle size.


