Vision 2017-2022
The NIH Center for Macromolecular Modeling and Bioinformatics Looks Forward
24 April 2015
By Lisa Pollack

The Center for Macromolecular Modeling and Bioinformatics has been in existence in Urbana, Illinois since 1990. Funded by the National Institute of Health (NIH), the mission of such NIH Centers has been to develop cutting-edge technologies and disseminate them to the broader community of biomedical researchers. The technologies could be laboratory instruments, but in the case of the Urbana Center, it is software. For example, NAMD and VMD are two programs that have been in existence since the 1990s–both developed with Center funding; NAMD is the molecular dynamics code, and VMD is the visualization and analysis software–they are used by thousands of researchers worldwide. The mission of the Center as well is to train scientists in the use of its technology.
The NIH funds its Centers in five-year increments. As such, the Center for Macromolecular Modeling and Bioinformatics will submit a renewal in May 2016 to extend its work into 2022. The proposal will not only document the accomplishments of the Center, but mainly it will offer its vision for the direction of its technologies and research for 2017-2022. As such, Center members are already thinking about the future directions for their technologies as well as what driving research projects to focus on. The Center consists of PI Klaus Schulten, six co-PIs (Aleksei Aksimentiev, Department of Physics, Laxmikant Kalé, Department of Computer Science, Zaida Luthey-Schulten, School of Chemical Sciences, James Phillips, the lead NAMD developer, John Stone, lead VMD developer, Emad Tajkhorshid, The School of Molecular and Cellular Biology) and the software developers. To present the strongest possible proposal to the NIH, the team decided to preview the long-range (2017-2022) vision of the Center at the Advisory Board review meeting. Every year the NIH requires its Centers to consult with an Advisory Board, to give the community the chance to look inside the Center and comment on its ideas, plans, and accomplishments. As such, on 10 December 2014 the Center held a day-long meeting where developers and co-PIs presented their vision for the future of the Center. Schulten thought that previewing their long-range vision to the Advisory Board would provide constructive criticism from experts and hence allow the Center ample time to tweak its vision before the final proposal is due.
But what does the Center have on tap for the future? As their technology product for the community is software, what do they foresee are the up-and-coming trends and directions in computing that they need to take advantage of? What have their users indicated they want as new features in the software products? What should they do in the climate of increasingly larger amounts of data that need to be analyzed? How should they best train new users when funding is growing scarcer? Below is a summary of what the Urbana Center envisions it should bet on in a climate of rapidly evolving technologies.
The Overarching Vision
Klaus Schulten started off the day with an overview of the general themes guiding the future vision. The Center has spent the last five years tailoring its software to handle massive systems (up to 100 million atoms), and Schulten still believes accurate large-scale simulations will be important.

The HIV capsid. At 64 million atoms, a milestone system for the Center.
However, at the same time, Schulten sees a need to work on small and intermediate-sized systems. Many biological processes happen on the millisecond timescale, and Schulten wants to push the molecular dynamics software NAMD to reach this range.
Also, Schulten stressed that the Center wants to continue to invest in breakthrough technologies (exact details will be discussed below). The Center was one of the earliest users of GPUs, and it was recently announced by the Department of Energy that it will be buying next-generation supercomputers that are heavily GPU accelerated, so Schulten foresees more investment in these accelerators by Center developers. Some other areas of investment will be cloud computing, tablets, and remote visualization. As for algorithms, new advanced path sampling techniques should aid the quest to reach longer and longer time scales, and incorporating quantum chemical forces should improve accuracy.
Finally, Schulten stressed that the Center will continue to collaborate with bioengineers to aid in their nanosensor development efforts. Schulten's group has already worked with many engineers at the University of Illinois on collaborative projects on biosensors. These opportunities will only multiply now that a new bioengineering-based medical school is planned for the Urbana campus, to come online, incidently, right in 2017.
NAMD–Nanoscale Molecular Dynamics
Sanjay Kalé is a computer scientist expert in parallel programming, who has been with the center since 1991, and whose guidance was critical to the lasting success of NAMD. Kalé considers NAMD a "bleeding edge" technology. That is, it is one step beyond cutting edge. Part of the reason for calling NAMD bleeding edge is because this software product has tried to stay ahead of computer hardware technology. For example, NAMD was very early in its embrace of parallel computers as well as GPUs (graphics processing units, sometimes known as accelerators).

Kalé feels that all the new hardware slated to come out could offer huge opportunities for NAMD. Some dramatic changes to hardware technology will be in products like processors, memory, and accelerators (like GPUs or the Xeon Phi). The DOE has already commissioned a program (DOE Fast Forward) to look into the hardware that needs to be in development now for exascale computing by 2022. While all these hardware advances are opportunities, they are challenges at the same time. But NAMD should not only focus on embracing hardware innovations–exascale operating systems as well are in the works. One area that NAMD could focus on here is power considerations; vastly more powerful machines will require vastly more electricity, translating to tens of millions of dollar a year at an exascale computer center just to pay the light bill. Reducing power consumption needs via software is one way to help address this mammoth issue of a power-hungry supercomputer, and an area where NAMD can lead the way.
Jim Phillips, lead NAMD programmer since 1998, envisions for 2017-2022 "Biomolecular Simulation Without Boundaries." Namely, whatever the current boundaries are on biomolecular simulation, NAMD should push back on them to enable scientists who use this tool to go wherever they want. One thing on the horizon is to develop tighter integration between NAMD and VMD, making it possible to do VMD-like analysis also in NAMD, and vice versa. Another goal is to reach more relevant time scales in molecular dynamics on a desktop, such that Phillips has set as a target a microsecond per day for 100,000 atoms on one node. But there is also a role NAMD could play in cloud computing, as Phillips foresees the cloud becoming a popular way to have powerful clusters on campuses. Phillips imagines using a tablet to do a NAMD interactive simulation while the cloud actually runs the calculation. On the other end of the computing power spectrum, NAMD will be focusing on large (over 100 million atoms) systems on the newest powerful supercomputers slated for debut in 2018. NAMD will be readying itself for exascale grand challenges by running large jobs on these 100 petaflop machines. And finally, in addressing the issue of accuracy, there are myriad improvements Phillips can focus on in NAMD with force fields, related to things like protonation, polarizability, and QM/MM, to name just a few.
VMD–Visual Molecular Dynamics
John Stone, lead developer for VMD, the center's visualization and analysis software, is excited about all the new technological developments that will allow a remarkable expansion of VMD capabilities for the 2017-2022 phase. One such ensuing feature is multi-viewport displays, where the user can have many windows open at the same time, for example, looking at four different angles of the same large molecule.

Lead VMD developer John Stone holds a pair of goggles that allow 3D viewing on a touch pad screen.
Stone talked about the myriad possibilities for VMD to utilize the up-and-coming user interface technologies. Imagine VMD running on a phone and viewing on the phone's screen a molecule in 3D just by wearing a head-mounted pair of glasses, or looking at a stereoscopic image of a virus on a tablet. This kind of viewing experience wouldn't be limited to phones or tablets, but could also be extended to desk surfaces, smart TVs and high-resolution monitors. Furthermore, continuing its tradition of interactive molecular dynamics in VMD, Stone would like to see this feature expand, and exploit all the ways users could manipulate their molecules on a touch screen of a tablet.
While many think of VMD as focused on visualization, the developers have come to regard it as an analysis tool as well. And Stone wants to see these analysis features become much more interactive. For example, trajectory analysis is done in an all-or-nothing way right now, but Stone envisions making this feature as seamless as in Google Maps, where you can do your analyses by zooming in or out with your mouse. The computing power of GPUs should allow such computations to be calculated on the fly. And lastly, in the future, Stone predicts VMD will need to handle systems with over one billion particles. Right now VMD's limit is right around the one billion mark, but Stone is confident that VMD can cross that milestone.
MDFF–Molecular Dynamics Flexible Fitting
The methodology MDFF is a relatively recent addition to the Center's toolbox, debuting circa 2008. The idea behind MDFF is that it functions as a means to fit high-resolution data from X-ray crystallography into lower-resolution electron microscopy maps. Developed initially for the ribosome, a large molecular machine, MDFF works especially well for large structures and is based on NAMD and VMD. This gives MDFF a powerful baseline, for users of MDFF can take advantage of all the myriad features of both NAMD and VMD when fitting their data. In 2014 the center introduced a variant, xMDFF, which essentially fits a model into a low-resolution X-ray diffraction structure via iteration. The center has many plans for both MDFF and xMDFF for 2017 and beyond.
First, however, lead programmer for MDFF Ryan McGreevy answered the question: Is there still a role for MDFF in a climate of increasingly more high-resolution electron microscopy maps? The answer was a resounding yes. For one, there are often many washed-out areas in an electron microscopy map from flexible parts of a structure and MDFF can play a definite role there. And McGreevy also predicts that some structures, which may be large, complex, or flexible, may not yield high-resolution maps within the next 10 years but instead be lower resolution. While advances in electron microscopy are on the Center's radar, advances in X-ray crystallography will also be important to the method xMDFF. X-ray free electron lasers will become increasingly more widespread and used on traditionally difficult-to-crystallize structures. For example, the use of these X-ray free electron lasers on nanocrystals of membrane proteins will often produce low-resolution structures, and xMDFF can benefit from this explosion of data.
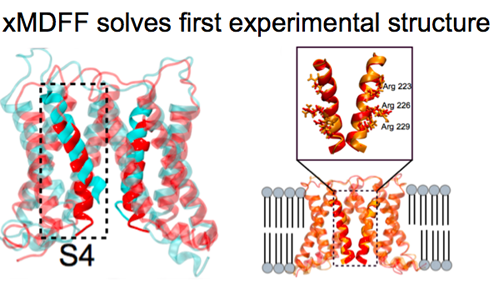
While xMDFF is poised to take advantage of experimental improvements, McGreevy foresees the need to bring xMDFF to crystallographers and thus expand the reach of xMDFF. For this he envisions interfacing to a popular crystallography package called Phenix. While Phenix already plays a core role in xMDFF, the Center anticipates that crystallographers could just use a Phenix interface to run xMDFF, and never have to interact with NAMD or VMD. Additionally, McGreevy would like to develop new crystallography tools within VMD that are based on Phenix's extensive toolbox, to further support xMDFF users.
But enhancements to xMDFF are not the only ones slated for 2017-2022. There are core improvements to MDFF that the Center wants to inaugurate. The result would be what McGreevy calls a "smarter, intelligent, adaptable MDFF." Imagine if, while NAMD is doing the fitting simulation, it could access information about the quality of fit in real time, and re-adjust parameters on the fly to improve the final product, all without extra user effort. That would save time, save computer resources, and make the best use of the time dedicated to running the simulations. The user wouldn't have to wait for results to readjust parameters, and then have to start everything all over from scratch. NAMD would intelligently adapt during its simulation. McGreevy believes, for example, that GPU cross correlation (currently done through VMD) could be integrated into NAMD and that all the information gleaned from it would make the MDFF fitting process smarter.
Finally, the center wants to tackle a problem common to molecular dynamics users and especially to users of MDFF: missing pieces of a structure. Before a flexible fitting simulation can even be run, users first have to turn to outside modeling software (like Modeller or Rosetta) to fill in the gaps in their structures. Why not just make an interface in VMD to these modeling software packages so that the user has everything at their fingertips? VMD would not only interface to modeling software, but also conveniently display scoring functions from the external software to help MDFF users make better decisions about which candidate models to choose. On top of this feature, if users are missing an entire high-resolution structure altogether, the center wants to fully integrate the structure-prediction tool MUFOLD (developed by the Dong Xu group at the U. of Missouri) into MDFF, so that users don't have to keep switching back and forth between MUFOLD and VMD/NAMD all the time. Ideally, MUFOLD does the prediction and analysis, corrects itself, and produces models that have already taken the density into account, and users would just see the final candidates in VMD, without needing to learn a whole other software package. The long-term aim here is to make MDFF a complete modeling package, and better anticipate the needs of its users.
Lattice Microbes–Whole Cell Simulation
The newest suite of programs at the Center is Lattice Microbes, an effort spearheaded by Professor Zan Luthey-Schulten, and first made available to biomedical researchers in 2012. This software can model an entire cell, or even colonies of up to a million cells.
Tumor Growth and Angiogenesis, a future project for Lattice Microbes.
One example of increasing complexity involves yeast. Luthey-Schulten discussed how this organism is becoming more and more important as a model organism, for about sixty percent of the 6000 yeast genes have human homologues. Thus, as experimentalists are increasingly studying it, more experimental information is coming out every day that Lattice Microbes can utilize, which will only improve the simulations for yeast. With more experimental input data, Luthey-Schulten hopes to add layers of complexity into her template of this eukaryotic cell, such as more compartments and more reactions. This will benefit the outside researchers who wish to use Lattice Microbes, for Luthey-Schulten releases template systems for use as a starting point.
But yeast is not the only system of interest in Lattice Microbes. For 2017-2022, Luthey-Schulten plans to work on models for biofilms, stem cells, cell signaling, and tumor growth. Additionally, she will be studying in vivo assembly processes, for example, ribosome self-assembly in a crowded cell. As mentioned above, more and more experimental data come out every day. Luthey-Schulten wants to make better use of systems biology data, such as fluxes, RNA sequence data, and enzymatic data to name just a few. However, to add more kinetic data, means to add more parameters. Hence there is a need to focus on the best way to automate parameter development so it is a seamless experience for the user. And finally, Luthey-Schulten wants to build more realistic models of the membranes she uses in Lattice Microbes.
Brownian Mover and Membrane Environment Modeler
While all-atom molecular dynamics is a mainstay of the Center, co-PI Alek Aksimentiev (whose vision was presented to the advisory board by postdoc Chris Maffeo) has his sights set on complex systems just beyond the length scales that molecular dynamics can reach. An example of this is the DNA replication fork, which is the complex of proteins that unwinds and then copies DNA in the nucleus. Such large cellular assemblies are a prime target for modeling, and Aksimentiev conjectures a coarse-grained approach can do the job, namely, reach biologically relevant timescales otherwise unavailable to the modeler. To accomplish this, Aksimentiev envisions both a direct coupling between different all-atom simulations, and a coarse-grained simulation running in parallel. He believes he can incorporate this method into NAMD and even use VMD for parameter development, thus building on the Center's existing tools.

Emad Tajkhorshid discusses the immense complexity of compartments walled off by membranes.
Professor Emad Tajkhorshid's area of expertise is membrane proteins and membrane-associated phenomena. One subject he is working on now is large-scale structural transitions in membrane transporter proteins. When membrane transport proteins, which are found all over the body, actively transport substances across a membrane, they often undergo huge conformational changes; knowing how they move during their function might pave the way to developing pharmaceutical drugs to work on the intermediate structures. Looking into the future, Tajkhorshid sees many opportunities in this rich field of research that is only expanding as more structures of membrane transporters are being solved. Tajkhorshid wants to develop tools and methodologies to elucidate these transporters. Specific areas to target include enhanced sampling, replica exchange, and collective variables. Tajkhorshid was clear to point out that large-scale structural changes can be applied to other proteins and other biomolecular processes, so that any new methodologies will have large appeal.
Finally, Tajkhorshid came back to a major theme of the meeting, which is the goal of whole cell simulation. A very important aspect of an entire cell is its many membranes, which add layers of complexity to the task of simulation. Hence, looking to 2017-2022, Tajkhorshid proposes Membrane Environment Modeler. This would be a comprehensive, versatile, and user-friendly suite for modeling membrane phenomena. He envisions tools that would allow a user to generate different structures, mutate lipids, or even conveniently change the composition of the lipids. He sees a need to have this tool kit to put together the very elaborate components of an entire cell.
Training and Tutorials
The center has taken an active role in training others to use its software products, be it the manifold online tutorials and case studies found on its website, or the hands-on workshops that have given one-on-one guidance to over 1,200 participants since 2003. While these aspects of training will certainly continue for the foreseeable future, the center has exciting new plans in its sights for 2017-2022, according to Danielle Chandler, assistant director of research in the Theoretical and Computational Biophysics Group.

Lead NAMD developer Jim Phillips discusses how NAMD is now running on tablets and iPhones via the Molecules App.
In 2014, through a lucky coincidence of events that could only happen in a small town, the NAMD developers teamed up with a local chemist and author Theodore Gray to help him realize a dream he had for the book he had just written called Molecules. Gray wanted to have a feature on the eBook version of Molecules that allowed readers to actually get a feel for how molecules behave. Enter NAMD, the perfect tool for describing molecules. What resulted was the "Molecules App." Available for the iPad and iPhone, the NAMD engine underlies this app, and users can literally manipulate over 300 small molecules with their hands using the touch screen of their devices. One can stretch or rotate molecules, or even tie them in knots, pulling up to 11 points at once.
The successful project, of translating NAMD's interactive molecular dynamics to a touch screen, has inspired the Center to think about how it could reach a broad audience if it could create interactive training materials. Currently a user can work through case studies (for things like DNA or membranes or ion channels), but how much more effective would it be for users to have an interactive experience on their tablets with, say, an entire virus particle. The center would like to embed VMD and NAMD into its own eBooks so that readers could literally play with the system with their hands. In this way, more people would become excited about the vast capabilities of VMD and NAMD, and perhaps the Center could even reach a younger audience.
Along the same vein, of broadening its audience base, Chandler also talked about how the prodigious training material database could be used in courses currently being taught at the University of Illinois. Some professors, like Klaus Schulten and Zan Luthey-Schulten, already teach courses with NAMD and VMD components built in. But the Center has plans to integrate its tutorials into existing courses; for instance, examples from computational biology could be used as illustrative case studies for traditional theory courses like non-equilibrium statistical mechanics. Or the Center even foresees working with departments to create new courses, as there is high demand at this university for computational biophysics skills.
Summary
Part of the mission of the Center is to push beyond the technological limits of modern computing. Use of state-of-the-art hardware combined with the Center's persistent software development has produced biomolecular simulation and modeling sophisticated enough to be called a computational microscope. In the past five years this microscope, facilitated by NAMD and VMD, has made possible elucidation of biological systems as large as one million to one billion atoms, thus making great leaps forward to describe the complex societies in a cell that are often made of aggregates of molecules. The Center's researchers and developers are therefore well poised to extend the reach of the computational microscope all the way to the whole cell in the ensuing five years; a driving principle here is to simulate a cell on the basis of physics.
But what benefits would there be to a computational microscope that can resolve immense complexes down to the atomic level? Living systems are extremely intricate molecular societies with many organizational aspects, but in essence they are all molecules. And a subtle molecular property has the potential to contribute to the function, or even dysfunction, of a complex biological organism. Already the Center has addressed problems like treatment of viral infections (1), the antibiotic resistance crisis (2), and preventing Alzheimer's disease (3). A long-cherished goal of biomedical research made possible with the Center's tools is to examine the societies of a cell to not only look for a molecular property out of balance (as in a pathology) but to also aid in treating pathologies with potential molecular candidates. The Center is using its combined wisdom, large assembled team, and proven track record to see that the computational microscope only grows more sophisticated into the year 2022.


