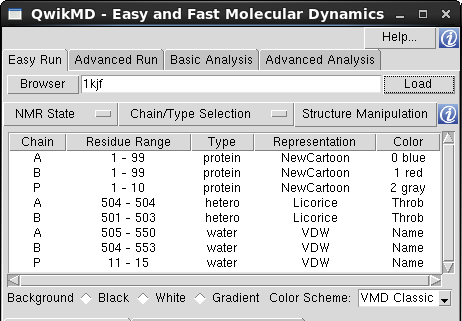
| Invoked from Easy Run |
Invoked from Advanced Run |
|
|
-
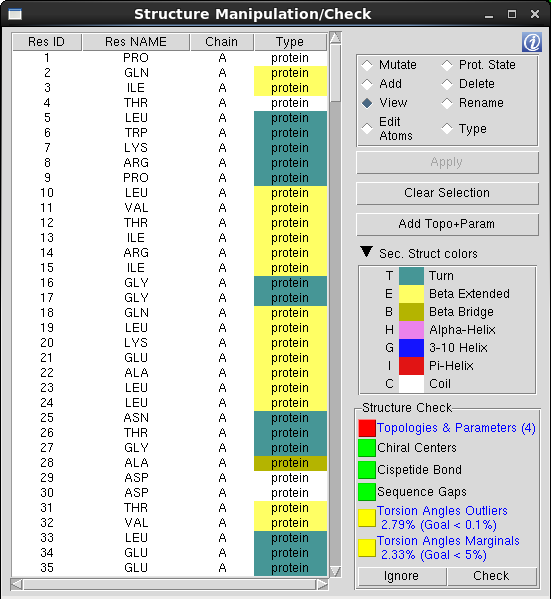
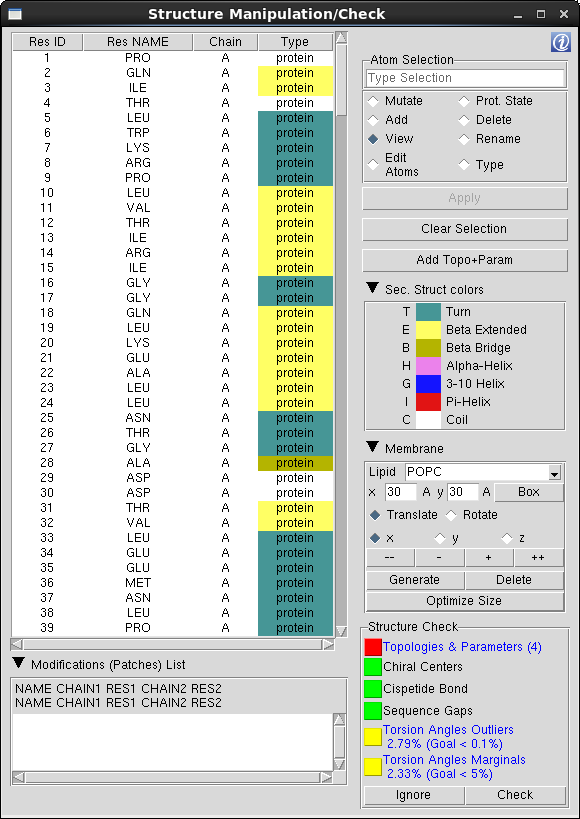
Residues Table: The Residues Table is main component of the "Structure Manipulation/Check" window and lists all the residues included in the selection made on "Chain/Type Selection" menu. The residues are listed in the same order as in the "Main Table", which is the order the residues are listed in the initial structure file. To facilitate the browsing through the residues list, one can change the sort criteria and sort the table by a specific column pressing the columns header.
Residues Marked in Red: When the residue is not recognized by QwikMD, meaning that the specific residue name doesn't match the residue names contained in the Topology files loaded by QwikMD. One must add the stream file containing the topology and the parameters, rename, change the residue type or delete the residue.
|
-
Table Options: The action triggered when a residue is selected on the Residues Table is determined by the table option selected. One should always select the table option first and then the residue(s) to be manipulated/visualized.
-
Mutate: Mark one residue to be mutated during the structure preparation phase. The mutations are only available for protein and nucleic residues and are dependent on the residue type, for instance, a protein residue can only be mutated to another protein residue.
WARNING: Even a very small number of mutations may affect the structure of a protein drastically. Be very careful when using the mutation tool of QwikMD as you might create artifacts in your simulation.
-
Prot. State: Mark one residue to assign a protonation state during the structure preparation phase. Amino acids, depending on their environment, can present different protonation states. It is recommended to check the protonation state of the amino acids before an MD simulation. Several tools can be used for that. One of the most popular tools is the PROPKA Server.
The assignment of protonation states is only available for protein residues and the options available are dependent on the residue selected, e.g., only two options are available for the aspartic acid (ASP), neutral (ASP) or protonated (ASPP) state.
Histidine Residues: Of the 20 amino acids, histidine is the only one that ionizes within the physiological pH range ~7.4. This effect is characterized by the pKa of the amino acid side chain. For histidine, the value is 6.04. This leads to the possibility of different protonation states for histidine residues in a protein, and makes the consideration of the proper state important in MD simulations. The viable states are one in which the delta nitrogen of histidine is protonated - listed with residue name HSD - one in which the epsilon nitrogen of histidine is protonated - HSE - and one in which both nitrogens are protonated - HSP. If not set by the user, QwikMD uses HSD as the standard histidine protonation state.
-
Add: Add residue(s) previously marked to be deleted.
-
Delete: Mark residue(s) to be deleted during the preparation phase.
-
View: Disable all manipulation options and only represent the selected residue(s).
-
Rename: Change the residue name to match the residue names in the CHARMM forcefield topology files. The possibilities available to rename the selected residue are dependent on the residue type, such as a protein residue can only be renamed to a residue categorized as a protein residue. If the proper residue name is not available, one must add the correspondent Topology+ParameterFiles.str, change the residue type and further rename or delete the residue.
WARNING: The structure preparation phase cannot start while the system contains unrecognized residues.
-
Type: Sometimes, molecules categorization can be misleading (or even nonexistent), which hinders the correct identification of residues. For instance, if the user intends to mutate the nucleotide Adenosine Triphosphate (ATP) by another nucleotide, it would be possible to mutate to a Guanine, Adenine, Cytosine or Thymine (or Uracil in RNA), as they share the same category as nucleic residues (nucleic). To avoid such structural errors, QwikMD gives the possibility to change residues type (category), so a logical choice can be made.
-
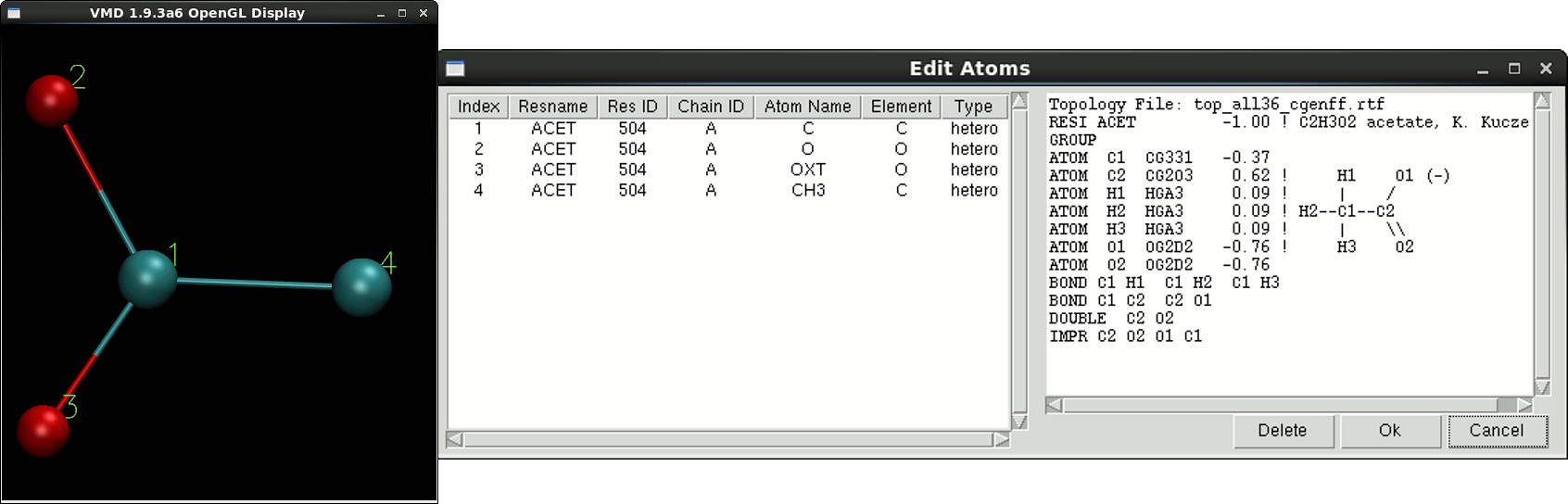
Edit Atoms: Residues fine grained edition, where one can change the residue number (resid), the atom's name and delete one or more atoms. Taking the example of the acetate residues in the HIV-1 protease structure (pdb entry 1KJF). The residue name in the original pdb structure is ACT and is marked as unrecognized by QwikMD since the same residue is named as ACET in the CHARMM topology file. In this case, one must change the residue name from ACT to ACET, which raises another topological issue, where none of the atoms name in the pdb match the atoms name the topology. In this case, one must rename also the atoms. For reference purpose, the topology entry and a numbered molecular representation of the residue is given to user.
|
-
Apply/Edit button: Apply residues selection during the SMD Anchoring/Pulling residues selection or open the "Edit Atoms" window when the "Edit Atoms" option is selected.
-
Clear Selection: Deselect the current selected residues.
|
|
Topology & Parameters Selection
Top
|
|
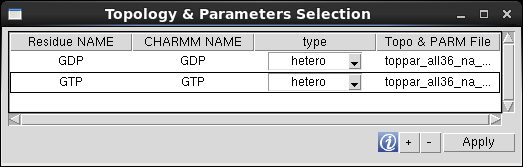
QwikMD doesn't lists all the residues present in the CHARMM 36 topology files by default for simplicity purposes, due to the vast list of residues available. If one needs to add more standard residues to QwikMD list, such as, proteins, nucleotides, carbohydrates (glycan), lipids or other standard residues, one must upload only the topology file (if the parameters are already included in the files loaded in QwikMD by the default) correspondent to the residue type and select the residue from the table.
If the system to be prepared includes one or more residues requiring special topologies and parameters, one must add a stream file (*.str) containing both residue's topology and parameters information.
-
Default Topology files:
- top_all36_carb.rtf, top_all36_cgenff.rtf, top_all36_hybrid.inp, top_all36_lipid.rtf, top_all36_na.rtf, top_all36_prot.rtf
-
Default Stream files:
- toppar_all36_carb_glycopeptide.str, toppar_water_ions_namd.str
-
Default Parameters files:
par_all36_carb.prm, par_all36_cgenff.prm, par_all36_lipid.prm, par_all36_na.prm, par_all36_prot.prm
-
To add the information regarding a special residue, one must press the "+" button and select the *.str file. QwikMD analyzes the file and list the residues ("RESI" keyword) and stores a copy in the QwikMD library folder.
-
Residue NAME: name to be displayed in QwikMD residues list. Protein and nucleic residues must have the same "Residue NAME" and "CHARMM NAME", while hetero, glycan and user defined residue types can assume different residues names, such as, the acetate ion residue: "Residue NAME"=Acetate", "CHARMM NAME"= "ACET".
-
CHARMM NAME: residue name in the CHARMM topology file. Note: Sometime the residues' name listed in the CHARMM topology files can be composed by more than 4 characters, which is a violation of the pdb file format. In this case, a warning message is prompted to the user and the correspondent line of the residue colored in red and one must rename the "CHARMM NAME" to a unique 4 letter residue name. The name of the residue is also updated in the file stored in the QwikMD library folder.
-
Default Parameters files:
par_all36_carb.prm, par_all36_cgenff.prm, par_all36_lipid.prm, par_all36_na.prm, par_all36_prot.prm
-
-
Topo & PARM File: File containing the residues topology and parameters.
WARNING: After all the editions to the residue's list and before leaving the "Topology & Parameters Selection" window, one must press "Apply" and reset QwikMD gui by pressing the "Reset" button in one of the Run Tabs.
|
|
Structure Check
Top
|
|
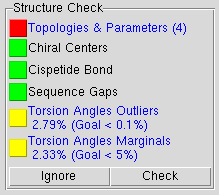
QwikMD checks the initial structure for structural inconsistencies in 4 different tests:
-
Topologies & Parameters: Comparison between the residues' name in the initial structure and the residues' name in the CHARMM36 topology files. Once a mismatch is detected, the residue is marked in red in the "Residues Table" and the correspondent chain is colored as "Throb" in the "Main Table". The system preparation phase cannot start before this warning is solved.
-
The easiest way to overcome the lack residue topologies and parameters is to delete the residue, which may not be desirable as the referred molecule might well be relevant. Thus, one has to provide QwikMD with the missing force field topology and parameters. If the topology and the parameters are available, i.e., in the literature, but not yet added to the force field, the user may add to QwikMD through Topology & Parameters Selection. These parameters can also be obtained from web servers, with varying levels of accuracy, such as CGenFF webserver. The most accurate and recommended method for furnishing missing parameters requires advanced knowledge, namely familiarity with the Force Field Tool Kit (FFTK) plugin in VMD. The ffTK plugin assists with the process of parameterization, employing quantum chemistry programs.
-
Chiral Centers: Detection of cis chirality errors in protein and nucleic acid structures using the Chirality plugin in VMD. For more information how to fix chirality errors please follow the Structure Check tutorial.
-
All amino acids but glycine have at least one chiral center at Cα. Threonine and isoleucine have an additional chiral center at Cβ. According to the D- / L- naming convention, naturally occurring amino acids are found in the L-configuration. Note, however, that D-amino acids do occur in biology, e. g., in cell walls of bacteria. Nucleic acids also have chiral centers. For example, in DNA the atoms C1', C3', and C4' are chiral, while RNA has an additional chiral center at C2'. Chirality is central to all molecular interactions in biological systems. A simple experiment demonstrates the principle: try to shake someone's left hand with your right.
-
Cispetide Bond: Detection of cis peptide bonds in protein structures using the Cispeptide plugin in VMD. For more information how to convert fix chirality errors please follow the Structure Check tutorial.
-
In naturally occurring proteins most peptide bonds are in the trans configuration. However, sometimes cis peptide bonds do occur. The vast majority of cis peptides is observed at a proline, Xaa-Pro, Xaa being any amino acid. But non-proline Xaa-non Pro cis bonds are also found in proteins, although they occur much less frequently than Xaa-Pro. The configuration of the peptide bond is central to the sort of secondary structure the protein backbone can adopt. It is easy to understand by imagining a normal α-helix and converting a trans peptide bond into its cis form. The result is that the hydrogen bond network stabilizing the helix is broken and the helix will be unstable in long simulation.
-
Sequence Gap: Detection of non-consecutive numbering of the protein and nucleic residues within each chain. If the residues sequence present a gap, another test is performed to evaluate if the residues are bonded, avoiding false detection of pre-crystallization sequence deletions. Due to the complex structure of carbohydrates (glycan), this test is not performed for such molecule types.
-
PDB Insertion Codes: Sometimes the initial structure presents insertion codes in the residues sequence. Rather than be numbered sequentially, some structures present residues numbering as 47, 47A, 47B and so on. This different numbering rises from the comparison between homologous proteins which have different chain lengths, and the authors who wants to preserve the reference among the different proteins use additional characters to identify non-matching residues between homologous sequences.
QwikMD reorders the residues to avoid unintentional deletion of the residues presenting different insertion codes. During the loading process, the residues sequence is checked for the existence of insertion codes and new residues IDs are assigned to ensure sequential numbering. In the end of the check structure process, a warning message is prompted to the user followed by the reference table displaying the relationship between the initial and the new numbering sequence. The reference table is then saved in the "setup" folder, within the working directory during the preparation phase, in the file "Renumber_Residues.txt".
-
Torsion Angles Outliers/Marginals: Evaluation of the distribution of the φ and ψ backbone angles. The percentage of angles consider as Outliers and Marginals are good indicators of the stereochecimal quality of the structure. QwikMD uses the command line of Torsion Plot VMD plugin to perform this evaluation. For more information, please visit the Ramaplot Plugin.
|
|
Membrane
Top
|
|
The "Membrane" field is activated when the Structure Manipulation/Check window is invoked from the Advanced Run. In here, one can defined the dimensions and position of the lipid bilayer. QwikMD uses the Membrane VMD plugin to generate the membranes.
-
Lipid: Membrane lipid composition. Only two types of lipid composition are currently available:
-
POPC: membrane 100% composed by 3-palmitoyl-2-oleoyl-D-glycero-1-Phosphatidylcholine
-
POPE: membrane 100% composed by 3-palmitoyl-2-oleoyl-D-glycero-1-Phosphatidylethanolamine
-
x/y entries: membrane dimensions in the x-axis/y-axis in Å. The initial position of the membrane is assigned to the center of the system currently selected.
-
Box: Draw the box where the membrane will be positioned.
Note: Due to the slow process of building the membrane and to avoid rounding floating point arithmetic rounding errors generated during coordinates translation and rotation process, the position of the membrane is defined using a temporary box, and then the membrane is generated once. If one decides to change the position of the generated membrane, first the membrane must be deleted pressing the "Delete" button, reposition the box, and generate the membrane again.
-
Translate/Rotate: the action generated by the movers "--", "-", "+" and "++".
-
x / y / z: axis for translation/rotation.
-
-- / ++ buttons: membrane temporary box movers to translate the box by -5 Å or +5 Å, or rotate -15° or +15° over the selected axis.
-
- / + buttons: membrane temporary box movers to translate the box by -1 Å or +1 Å, or rotate -1° or +1° over the selected axis.
-
Generate: Generate the membrane according to the location and dimension of the temporary box.
-
Delete: Delete the previous generated membrane.
-
Optimize Size: Change the center and dimensions (orientation is not affected) of the membrane to ensure that the existence of at least 15 Å between protein maximum and minimum coordinates and simulation cell limits in the membrane axis.
Note: The periodic boundary conditions dimensions, in the membrane axis, are determined by the membrane dimensions.
|
|
Modifications (Patches) List
Top
|

|
|
During the structure preparation, the initial structure is split into individual segments, such as, the segment containing all protein residues belonging to the chain A, protein residues of the chain B and the segment containing all water residues belonging to the chain A (see psfgen tutorial). Often times, special modifications (patches) are necessary after the creation of the segments, such as a connection between two different protein segments, or a crosslink between an Heme group and the protein chain.
One can apply structure modifications by listing the sequence of patches, following the specific command format, "PatchName ChainID1 ResidueNumber1" or in cases of 5 arguments patches "PatchName ChainID1 ResidueNumber1 ChainID2 ResidueNumber2". The list of modifications can be found in the CHARMM36 topology and stream files with the keyword "PRES".
|
|