Hybrid QM/MM NAMD
NAMD QM/MM interface extends existing NAMD features to the quantum mechanical level, presenting features that are not yet available in any QM/MM implementation. The first is the ability to execute multiple QM regions in parallel, thorough independent executions of your choice of quantum chemistry code. This allows one to account for multiple reaction centers that are known to work synergistically, for example, or even distant allosteric regulation sites and a reaction center. Investigation of processes occurring on a timescale usually not accessible by QM/MM methods can now be performed by a combination of temperature replica exchange molecular dynamics and QM/MM molecular dynamics. Another innovation comes from the fact that most QM/MM implementations have pre-defined QM and MM regions with no changes of atoms between these regions during a simulation, which would not efficiently allow the study of translocation of DNA in, e.g., nanosensors. Taking advantage of an easy-to-use Tcl based interface and capabilities integrated from VMD, NAMD QM/MM allows the update of the QM and MM regions at every step of the QM/MM simulation.
Recent News and Announcements
Team brings subatomic resolution to computational microscope
Scientists have built a "computational microscope" that can simulate the atomic and subatomic forces that drive molecular interactions. This tool will streamline efforts to understand the chemistry of life, model large molecular systems and develop new pharmaceutical and industrial agents, the researchers say. read more
NAMD 2.12 Release
NAMD 2.12 is now available allowing hybrid QM/MM calculations. More information will be available soon.
VMD QM/MM Features
The new VMD, to be realeased in early 2018 (alpha version already available), contains many QM/MM featues, incluing new orbital representations and simulation setup and control through the QwikMD interface. More information will be available soon.
Availability
Hybrid QM/MM calculations are a new feature available in NAMD 2.12 and newer.
Download NAMD 2.12 for free here. We recomend the use of the nightly build version as the QM/MM interface is in constant development.
Hybrid QM/MM features are now available in VMD.
Download VMD 1.9.4 alpha version, with full QM/MM support, for free here. We recomend using the latest release as the QM/MM interface is in constant development.
How to cite
NAMD goes quantum: An integrative suite for hybrid simulations. Melo*, M. C. R.; Bernardi*, R. C.; Rudack T.; Scheurer, M.; Riplinger, C.; Phillips, J. C.; Maia, J. D. C.; Rocha, G. D.; Ribeiro, J. V.; Stone, J. E.; Neese, F.; Schulten, K.; Luthey-Schulten, Z.; Nature Methods, 2018 (doi:10.1038/nmeth.4638)
NAMD goes Quantum - YouTube
Introduction to hybrid QM/MM simulations
This brief introduction aims at providing some basic context to the following description of capabilities and commands available in NAMD's QM/MM interface.
- Division of Labor
The basic idea behind a hybrid QM/MM simulation in NAMD is to use a classical force field to treat the classical atoms in the system (or "MM atoms"), and pass the information that describes the quantum atoms in the system (or "QM atoms") to a Quantum Chemistry (QC) software, which is expected to produce gradients for all QM atom, as well as the total energy of the QM region (and optionally partial charges). All bonded and non-bonded interactions among MM atoms are handled by NAMD's force field. Similarly, all bonded and non-bonded interactions among QM atoms are handled by the QC software in its chosen theory level. Treatment of covalent bonds between QM and MM atoms will be described in a following section.
The non-bonded interactions between QM and MM atoms are handled differently, and can be modified and regulated by the user. Van der Waals interactions are always calculated, and can be done using either the default force field parameters, or specific (user defined) parameters for QM atoms. Parameter modifications for QM atoms have been proposed in order to compensate for over-polarization that these atoms may exhibit in hybrid QM/MM simulations. Larger Van der Waals radii and/or shallower well depths should then be provided for all element types that occur among QM atoms (see the "qmVdWParams" keyword).
- Mechanical and Electrostatic Embedding
Electrostatic interactions between QM and MM atoms deserve a more detailed discussion due to the abundance and diversity of available alternatives. The first decision to be made is whether there will be electrostatic interactions between the two portions of a system, QM and MM. In the "mechanical embedding" scheme, only positions and elements of atoms in the QM region are passed on to the chosen QC software for energy and force calculations. This way, QM and MM atoms share only Van der Waals interactions.
In the "electrostatic embedding" scheme, on the other hand, the partial charges of MM atoms surrounding all QM atoms are used to approximate the electrostatic environment where QM atoms are found (the schemed is selected with the "qmElecEmbed" keyword. See Figure 1. This process can be customized in a variety of ways, the first of which is deciding if a smoothing function will be used to avoid an abrupt decay in electrostatic force due to the cutoff used in the selection of surrounding point charges (this option is activated with the "qmSwitching" keyword).
Classical point charge utilization can be further customized by choosing which smoothing function will be used, and if the total charge of selected partial charges should be modified to (A) have a whole charge or (B) have a complementary charge to that of the QM region, so that the sum of charges from QM atoms and classical partial charges add to zero (see Figure 1).
With electrostatic embedding, QM atoms are influenced by the charges in the classical region. In order to balance the forces acting on the system, NAMD uses partial charges for the QM atoms to calculate the electrostatic interaction with classical point charges. There are two possibilities for the origin of the QM partial charges: the original partial charges found in the force field parameter files can be used, or updated partial charges can be gathered at each step form the QC software output (controllable through the "qmChargeMode" keyword). The continuous update in charge distribution allows for a partial re-parameterization of the targeted molecule at each time step, which can lead to an improved description of the interactions of a ligand as it repositions over the surface of a protein, for example, or as it moves through a membrane.
In case PME is activated by the user, NAMD will automatically apply the necessary corrections to handle the QM region, allowing it to be influenced by long range interactions from the entire system.
- Covalent Bonds Divided by the QM/MM Barrier
Hybrid QM/MM simulations of biomolecular systems often present situations where only a portion of a molecule should be treated quantum mechanically, usually to save computational resources since the cost of simulating QM regions rises rapidly with the number of simulated toms. In order to deal with chemical bonds that are split by the QM/MM division of the biomolecular system, that is, bonds that have one atom in the quantum (QM) region and another in the classical (MM) region (we will call these "QM/MM bonds"), NAMD makes approximations to the molecular system in order to bridge differences in simulation type (QM vs MM), and minimize errors involved in the QM/MM division of the system (Figure 2A and B).
- Link Atoms
As previously mentioned, the information regarding atoms in the QM region is passed on to the chosen QC software, that is, their respective positions and element types, but in order to maintain (or approximate) the effect of the chemical bond between the QM atom and the MM atom, NAMD creates and places a link atom (usually a hydrogen) along the "broken" QM/MM bond. The user can fine-tune this process by choosing the method of placement of the link atom and even the element of such atom (keywords "qmBondDist" and "qmLinkElement").
The introduction of the link atom will invariably place it very near the classical atom involved in the QM/MM bond, therefore the use and placement of partial charges from classical atoms becomes highly relevant. Under the mechanical embedding scheme, the QC software only receives the atoms in the QM region and the link atoms created to approximate QM/MM bonds, so no manipulation of partial charges is required. On the other hand, usual QM/MM simulations are done under the electrostatic embedding scheme, in which case the partial charges of classical atoms involved in the QM/MM bonds and classical atoms directly connected to them require special treatment.
- Point Charge Alterations
Several methods have been proposed to handle this situation, and the QM/MM interface developed here implements the most widely accepted ones. One can be chosen using the "qmBondScheme" keyword (Figure 2C to G). In all implemented methods, the classical atom participating in the QM/MM bond (MM1 atom) does not have its partial charge passed on to the QC software, since this would create excessive repulsion (or attraction) on the link atom. This is, in fact, the entirety of the "Z1" method: ignoring the partial charge of the MM1 atom. Analogously, "Z2" and "Z3" ignore all partial charges up to MM2 and MM3 atoms, respectively (Figure 2C to E).
The Redistributed Charge and Dipole (RCD) method (Figure 2F) is more elaborate, as it rearranges the partial charge of the MM1 atom (indicated as q) so that the total charge of the region is maintained as well as the dipole moments of the bonds between MM1 and MM2 atoms. This is done by creating "virtual" point charges, which are passed on to the QC software as if they represented partial charges of classical atoms. More specifically, the RCD method creates a virtual point charge in the middle of all MM1-MM2 bonds with a charge of 2*q/n, where n is the number of MM2 atoms connected to MM1, and also subtracts a charge q/n from each of the MM2 atoms, so that the total charge of the region remains constant while approximating the dipole moment of the MM1-MM2 bonds. This way there will be no point charge placed at the position of the MM1 atom, but its partial charge is not simply removed, it is redistributed.
A similar approach is taken by the Charge Shifting (CS) method (Figure 2G). In this case, the MM1 partial charge is equally distributed across the MM2 atoms, and two virtual point charges are placed along the direction of the MM1-MM2 bond, one before the MM2 atom and one after, each one with a charge of +q/n and -q/n respectively. This method will also keep the total charge of the region constant while trying to preserve the local dipoles formed by all MM1-MM2 bonds.
Along with the gradient over all QM atoms, NAMD can also use the partial charge derived from the QC calculation to update the charge distribution of atoms in the QM region. When a QM/MM bond exists, however, part of the charge of the region will be placed on the link atom, and in order to keep the charge of the QM region constant, the link atom charge is re-distributed on the QM region. This seemingly simple mechanism can cause problems unless special care is be taken when deciding which bond will mark the division of QM and MM regions.
Many force fields divide the topologies of biomolecules in "charge groups" (Figure 3A and B). What this means is that not only will the partial charges of all atoms of a molecule add up to the whole number that represents the charge of the molecule, they will also add up to whole numbers in sub groups of atoms (look for the words "GROUP statements" in this link to see an example). Therefore, one needs to make sure that the chosen QM/MM bond(s) sits in between "charge groups", so the total sum of partial charges of atoms defining a QM region is a whole number. This is especially important in order to keep the total charge of the system constant. Since the QC calculation will always distribute a whole charge over all atoms of a system (QM atoms plus a link atom), if the partial charge of QM atoms is not initially a whole number, it will be forced into a whole number after the first QC step, where the charge of the link atom is distributed over the QM region. This will create a mismatch between QM and MM charges, changing the total charge of the entire system (QM plus MM regions).
An example can be seen in Figure 3. If bonds 1 and 3 are chosen as the QM/MM bonds, the charge distribution seen in Figure 3C shows a whole charge for the QM region (and consequently for the MM region). Therefore, any charge placed on link atoms can be redistributed to the QM atoms with no change in total system charge. However, if bonds 2 and 3 are chosen for the QM/MM bond (Figure 3D), the charge of the MM region would be +1.16, while the charge of the QM region would be -1.16. Since the QC calculation would place a pre-determined whole charge on the region (-1, in this case), the updated charge of the QM region after the first simulation step would change the total charge of the system to +0.16, in this example.
- Custom Quantum Chemistry Software
In order to offer the broad range of tools and technologies present in NAMD to all researchers who develop and/or employ specialized Quantum Chemistry tools, the QM/MM interface is prepared to utilize any QC tool that can be wrapped in a script that converts input and output files to specified formats. This flexible interface will improve development and testing of new tools, as well as their quick integration utilization in hybrid dynamics.
We currently provide python wrapper scripts for GAUSSIAN, TeraChem and Q-Chem, but other wrapper scripts can be generated based on these templates, in any other language, as long as they are provided to NAMD in an executable form. ORCA is natively supported, but we also provide a python wrapper script here with extended comments explaining the format in which NAMD will write data for the QC software, and the format in which NAMD expects to find the results.
Aiming at large macromolecular simulations that could take advantage of localized QM resolution, NAMD allows the user to set up multiple independent QM regions in the same molecular system. For example, one could study a multimeric complex that contains several active sites and have all active sites be calculated with a chosen QC software simultaneously (Figure 4). Each active site would be calculated independently of all others, by its own execution of the QC software, keeping the calculation cost low and without impacting the overall efficiency of the simulation, since all QM regions would be calculated in parallel.
Identifying the different QM regions and which atoms belong to each one of them can be simply accomplished in the input PDB file, or in a dedicated PDB file (keyword "qmParamPDB"). Since each region can contain different sets of molecules, their charges and multiplicities are indicated separately (see keywords "qmCharge" and "qmMult").
For simulations of large systems that are distributed across several computer nodes, one can control how many independent QM regions are calculated in each node. This would prevent large simulations from running out of memory if two or more large QM regions are placed in the same node (see keyword "qmSimsPerNode").
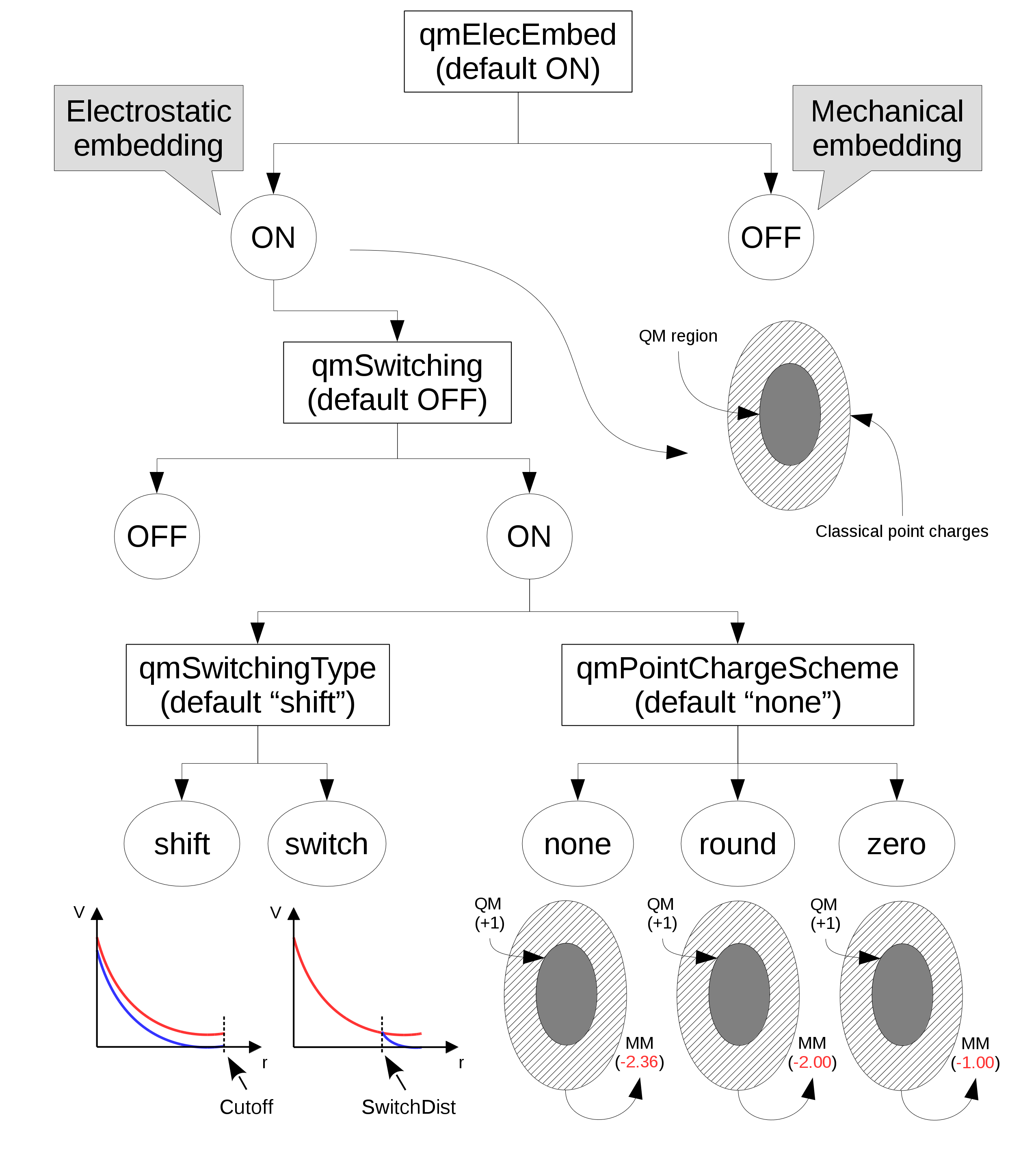
Figure 1: Diagram of options that control the use and manipulation of classical point charges. Default values are indicated below their respective keyword. "Cutoff" and "SwitchDist" are keywords used in NAMD to configure the calculations of electrostatic and Van der Waals interactions.
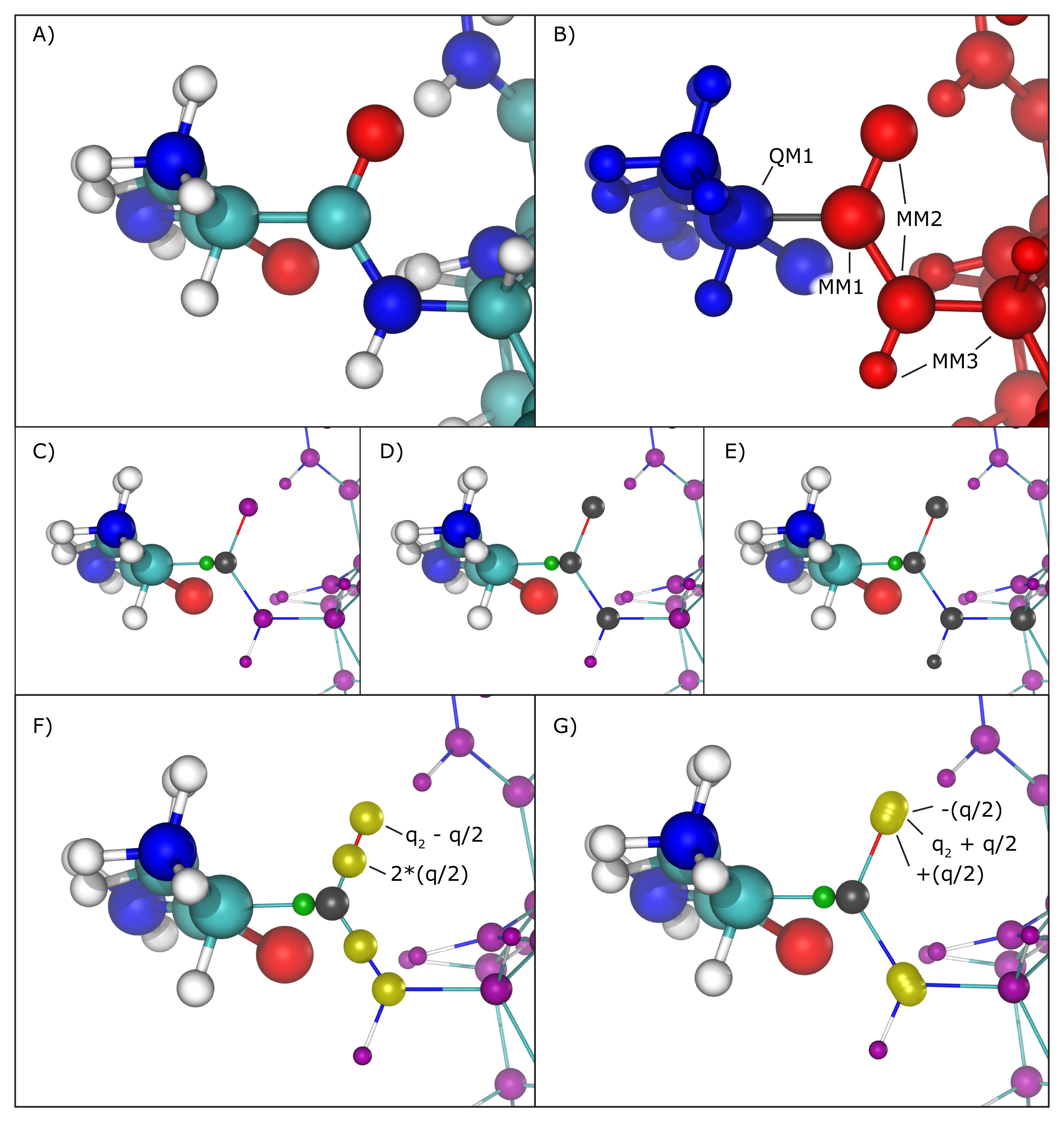
Figure 2: Treatment of QM/MM bonds. A) Illustration of all atoms in the vicinity of the QM/MM bond, colored by element: cyan for carbon, white for hydrogen, blue for nitrogen and red for oxygen. B) To the left, in blue, is the region that will be treated with the chosen QC software. To the right, in red, the region treated classically by NAMD. The bond in gray is the one crossing the QM/MM division. The atom marked as QM1 is the quantum atom directly connected to the classical atom on the other side of the QM/MM division. Analogously, the atom marked as MM1 is the classical atom directly connected to the quantum atom on the other side of the QM/MM division. Atoms marked as MM2 are directly bonded to the MM1 atom, and atoms marked MM3 are directly bonded to MM2 atoms. C) Z1 method. Ignored partial charges are indicated in the image with a gray sphere taking the place of its respective classical atom. Directly adjacent to MM1 is a green sphere representing the link atom that is placed along the QM1-MM1 covalent bond. All remaining partial charges representing classical atoms that are passed on to the QC software are indicated in purple spheres. D) Z2 method. E) Z3 method. F) RCD method. Virtual point charges, are represented in yellow spheres. The text indicates the total charge placed at each position, where q indicates the charge of the MM1 atom and q2 represents the partial charge of the MM2 atom at that position. The yellow spheres at MM2 atom positions indicate their partial charge has been changed from its original value. G) CS method. Since in this case the virtual point charges are placed very close to the MM2 atom position, the yellow spheres representing them show significant overlapping.
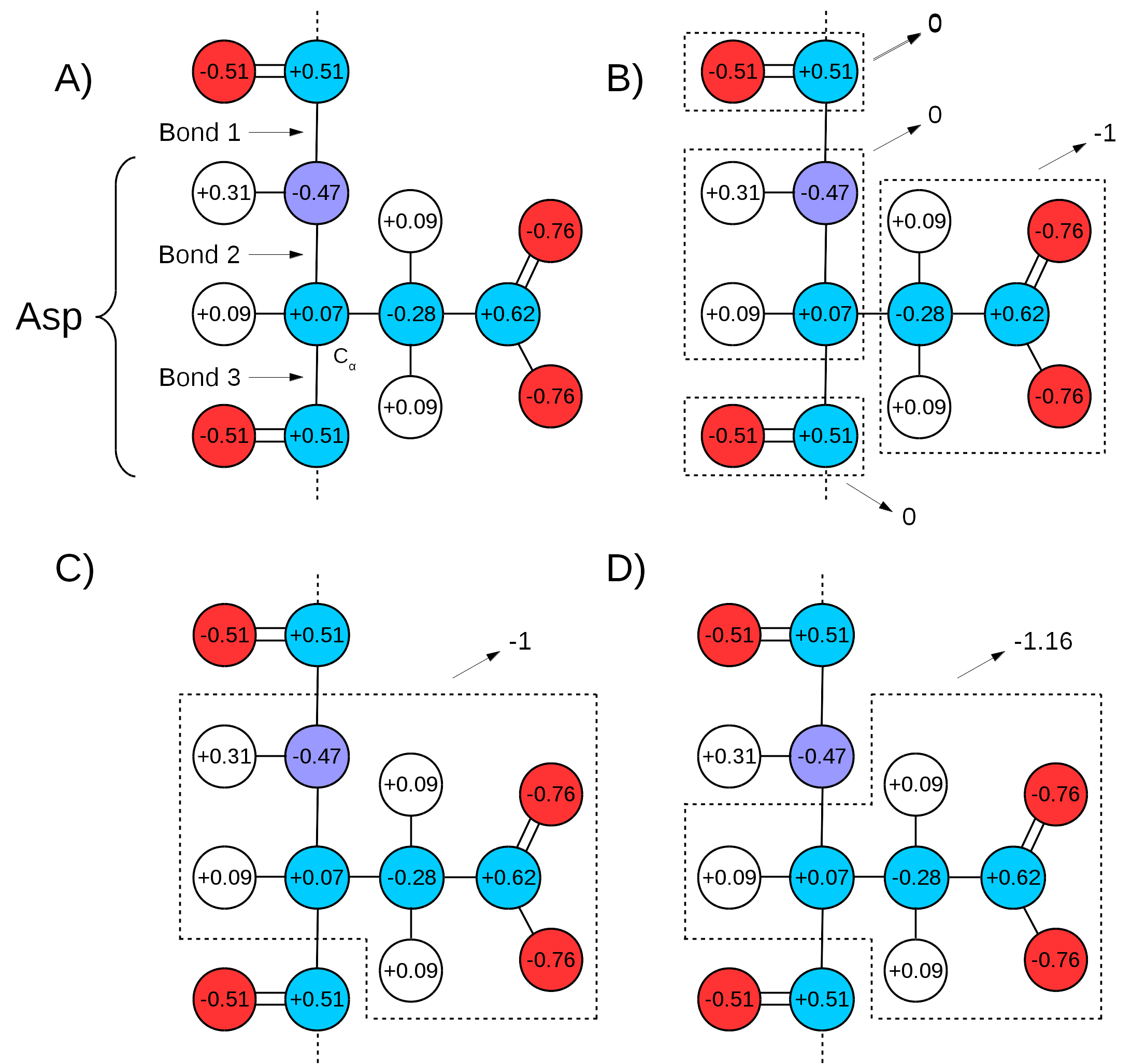
Figure 3: Charge Groups and QM/MM Bonds. A) Illustration of Aspartate and the distribution of charge over its atoms as in CHARMM36 force field parameters. Circles in red indicate oxygen atoms, blue indicate nitrogen atoms, cyan for carbon atoms, and white for hydrogen atoms. "Bond 1" indicates the peptide bond,"Bond 2" indicates the one between the alpha carbon and the peptide bond nitrogen, and "Bond 3" the bond between the alpha carbon and the peptide bond carbon. B) Charge groups are indicated with dashed squares, along with their total charges. C) Depiction of the atoms in the QM region if Bonds 1 and 3 are used to separate it from the MM region. The total charge of QM region is -1. D) Depiction of QM region if the same is defined by Bonds 2 and 3. In this case, the total charge of QM region is -1.16.
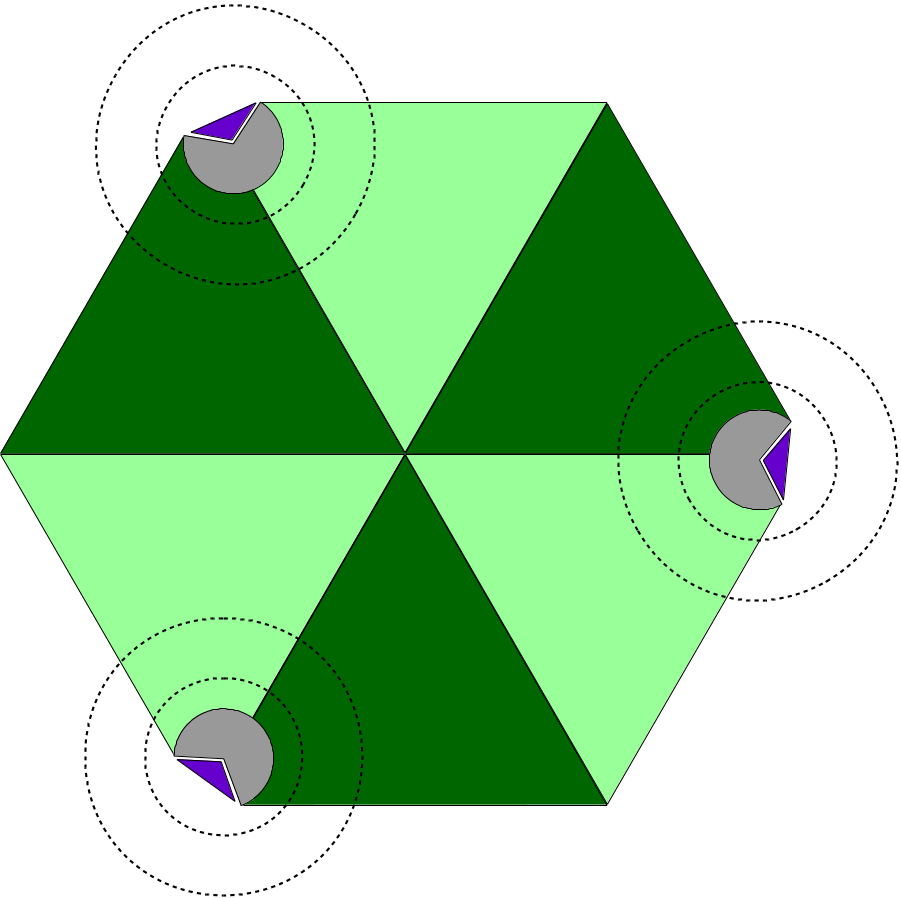
Figure 4: Diagram of Multiple QM Regions. The illustration depicts a hetero-hexameric complex (light and dark green triangles) that combine to create three active sites (in gray). Active sites are bound to a target molecule (purple triangle). The inner and outer dashed circles represent, respectively, the boundary of a QM region and the limits of the classical point charge shell around that region.
Keywords
qmForces
Description: Truns ON or OFF the QM calculations.
Acceptable Values: "on" or "off"
Default Value: off
qmParamPDB
Description: Name of an optional secondary PDB file where the OCCupancy or BETA column has the indications for QM or MM atoms. QM atoms should have an integer bigger than zero (0) and MM atoms should have zero as the beta or occupancy field. The same file may have indications for bonds between a QM atom and an MM atom. This should be provided in case the PDB file passed to the "coordinates" keyword already has data on its beta or occupancy columns, such as when a SMD simulations is being performed.
Acceptable Values: pdb file
qmColumn
Description: Indicates which column has the QM/MM field.
Acceptable Values: "beta" or "occ"
Default Value: required
qmSimsPerNode
Description: Number of independent simultaneous QM simulations per node.
Acceptable Values: integer
Default Value: 1
qmBondColumn
Description: Indicates which column has the QM-MM bond information. This will tell NAMD which atoms are at the ends of a covalent bond split by the QM/MM barrier, with one atom being quantum and one being classical.
Acceptable Values: "beta" or "occ"
Default Value: No default. If provided, NAMD will parse options for dealing with QM/MM bonds.
qmBondDist
Description: Indicates whether the value in the BondColumn will be used to define the distance between the QM atom and the Link Atom that will replace the MM atom in the QM system.
Acceptable Values: "on" or "off"
Default Value: off
qmBondValueType
Description: Indicates if the values in the BondColumn represent either: (LEN) the length between the QM and link atoms; or (RATIO) the ratio between the QM-MM distance and the one which will be used as the QM-Link distance.
Acceptable Values: "len" or "ratio"
Default Value: len
qmLinkElement
Description: User defined Link Atom element. Two syntaxes are allowed: if there is only one QM-MM bonds, a string with the element symbol is allowed (such as "H" or "Cl"). If there are two or more bonds, the string needs to have the two atoms that compose the bond, and then the element (such as "4 9 Cl"). The default element for all Link Atoms is hydrogen.
Acceptable Values: string (example: 4 9 Cl)
Default Value: H
qmBondScheme
Description: Indicates what will be the treatment given to QM-MM bonds in terms of charge distribution and link atom creation and placement. CS: Charge Shift Scheme; RCD: Redistributed Charge and Dipole method; Z1: Only ignored MM1 partial charge, with no charge redistribution; Z2: Ignores MM1 and all MM2 partial charges, with no charge redistribution; Z3: Ignores MM1 and all MM2 and MM3 partial charges, with no charge redistribution.
Acceptable Values: "CS" or "RCD" or "Z1" or "Z2" or "Z3"
Default Value: CS
qmElecEmbed
Description: Indicates if classical point charges should be used in QM calculations.
Acceptable Values: "on" or "off"
Default Value: on
qmSwitching
Description: This will scale down the point charges representing the classical system as to replicate the switching procedure that NAMD applies to all charged interaction (see "switching").
Acceptable Values: "on" or "off"
Default Value: off
qmSwitchingType
Description: This option is used to decide which kind of function will be used to scale down point charges sent to QM calculations. SHIFT: This will "shift down" the entire shell of point charges so that electrostactic interactions reach zero at the cutoff distance. SWITCH: This will only change point charges in the sub-volume between the switchdist and cutoff distance, so that electrostactic interactions reach zero at the cutoff distance.
Acceptable Values: "switch" or "shift"
Default Value: shift
qmPointChargeScheme
Description: This option allows the used to decide if and how the point charges presented to the QM system will be altered. NONE: Nothing will be done. ROUND: This will change the most distant point charges so that the total sum of point charges is a whole number. ZERO: This will adjust the most distant point charges so that the total sum of point charges is ZERO.
Acceptable Values: "none" or "round" or "zero"
Default Value: none
qmBaseDir
Description: This should be a fast read/write location, such as a RAM folder (/dev/shm on most linux distros). The user needs to make sure this folder exists in the node(s) running the QM calculation(s).
Acceptable Values: path to folder
#
qmReplaceAll
Description: Indicates to NAMD that ALL forces form NAMD will be ignored and only the gradients from the QM software will be applied on the atoms. This IS NOT NECESSARY in any regular QM/MM simulation, and will prevent the use of any other feature from NAMD such as SMD.
#Acceptable Values: "on" or "off"
#Default Value: off
#qmVdWParams
Description: The QM code will change all QM atom's VdW types to "q"+element (i.e. all carbons will be qC, all hydrogens will be qH) for VdW interactions. This means that a parameter file with epsilon and sigma values for these atom types must be provided along with the regular parameter files. For example, if using CHARMM force field, the new file should be in CHARMM's format.
Acceptable Values: "on" or "off"
Default Value: off
qmPCStride
Description: Sets a stride for new point charge determination. The same set of classical atoms will be sent to QM calculations as point charges, but with updated positions.
Acceptable Values: integer
Default Value: 1
qmCustomPCSelection
Description: Indicates that one or more file(s) will be provided with a custom selection of point charges. Each file will have a selection for a single QM group. This selection will be kept during the entire simulation.
Acceptable Values: "on" or "off"
Default Value: off
qmCustomPCFile
Description: The file will have, in the "qmColumn", the same QM ID provided for a single QM group. All other groups will have zero (0) in this column. In the second column (beta or occupancy), the classical or quantum atoms (from other QM regions) that need to be passed as point charges will be identified by a non-zero number.
Acceptable Values: pdb file
Example/Format:
QMCustomPCFile system/system.customPC.1.pdb
QMCustomPCFile system/system.customPC.2.pdb
QMCustomPCFile system/system.customPC.3.pdb
QMCustomPCFile system/system.customPC.4.pdb
qmLiveSolventSel
Description: With Live Solvent Selection (LSS), NAMD will automatically keep track of the solvent molecules for all QM Groups, and will exchange classical solvent molecules with QM solvent molecules every "QMLSSFreq" steps.
Acceptable Values: "on" or "off"
Default Value: off
qmLSSResname
Description: This indicate which residue name will be used in LSS.
Acceptable Values: resname (example: TIP3)
Default Value: TIP3
qmLSSFreq
Description: Frequency of LSS. Must be a multiple of steps per cycle.
Acceptable Values: integer (multiple of stepspercycle)
Default Value: 100
qmLSSMode
Description: For LSS, this indicates how solvent molecules are selected. In all cases, the closest solvent molecules are selected, and if a classical solvent molecule is closer than a QM one, they are swaped. DIST: This mode will use the smallest distance between a solvent atom and a non-solvent QM atom to sort solvent molecules. This is best used when the non-solvent QM atoms form irregular volumes (when the COM is not very representatve), and/or volumes with high solvent accessibility (such as a drug, or a small peptide, in solution). COM: This mode will sort solvent molecules based on Center Of Mass distance between the solvent COM and the COM of a selection for each QM group (see "QMLSSRef" keyword). Best used with small QM regions that have limited solvent accessibility, such as an active site.
Acceptable Values: "dist" or "COM"
Default Value: dist
qmLSSRef
Description: This will indicate which residues are to be used in the determination of the COM of non-solvent QM atoms. Only these atoms will be used to determine the closest set of solvent molecules. The keyword takes a string composed of the QM group ID, the segment name and the residue ID.
Acceptable Values: string
Example/Format:
QMLSSRef "1 RP1 9"
QMLSSRef "2 RP1 3"
QMLSSRef "2 RP1 2"
QMLSSRef "3 AP1 9"
QMLSSRef "3 AP1 3"
QMLSSRef "4 AP1 9"
qmConfigLine
Description: The string passed to "qmConfigLine" will be copied and pasted at the very begining of the configuration file for the chosen QM software if either ORCA or MOPAC are selected.
Acceptable Values: string
Example/Format (QM/MM NAMD-ORCA):
qmConfigLine "! PM3 ENGRAD"
qmConfigLine "%%output PrintLevel Mini Print\[ P_Mulliken \] 1 Print\[P_AtCharges_M\] 1 end"
Example/Format (QM/MM NAMD-MOPAC):
qmConfigLine "PM7 XYZ T=2M 1SCF MOZYME CUTOFF=9.0 AUX LET GRAD QMMM GEO-OK"
qmConfigLine "Test System"
qmMult
Description: Multiplicity of the QM region. This is needed for propper construction of ORCA's input file. Each string must be composed of the QM region ID and its multiplicity.
Acceptable Values: string
Example/Format:
qmMult "1 1"
qmMult "2 1"
qmMult "3 1"
qmMult "4 1"
qmCharge
Description: Indicates the charge of each QM region. If no charge is provided for a QM region, NAMD calculates the total charge automatically based on the given parameter set. Each string must be composed of the QM region ID and its total charge.
Acceptable Values: string
Example/Format:
qmCharge "1 1"
qmCharge "2 -1"
qmCharge "3 1"
qmCharge "4 -1"
qmSoftware
Description: Indicates which QM software should be used. In case the user wants to apply another QM code, this can be done by using the "custom" qmSoftware. In this case, NAMD will call the executable defined in the qmExecPath variable and will give it one argument: the full path to the input file.
INPUT: This input file will contain on the first line the number of QM atoms (X) and the number of point charges in the file (Y, which may be 0 or more), separated by a space. The following X+Y lines will have four (4) fields: X, Y and Z coordinates, and a fourth field which will depend on the type of entry. For QM atoms, the field will contain the element of the QM atom. For point charge lines, the field will contain the charge of the point charge.
OUTPUT: The expected output file whould be placed in the same directory as the input file, and should be named "*inputfile*.result" (meaning it will have the same path and name of the input file, plus the suffix '.result'). This file should have, on its first line, the energy of the system and the number of point charges that were passed to ORCA, and that ORCA calculated forces on (zero, if using mechanichal embedding). The two number should be separated by a single white space. Following the standard for the INPUT file, there will be another X+Y lines in the OUTPUT file. On the following X lines (where X is the number of QM atoms passed in the input file), there must be four (4) fields: the x, y and z components of the TOTAL FORCE applied on that atom, and on the fourth field, the charge of the atom. If the user indicates that charges from the QM software should not be used (see "QMChargeMode"), the fourth field should have zeroes, but should not be empty. On the following Y lines (where Y is the number of point charges), there must be only three (3) fields: the x, y and z components of the electrostatic force applied on that point charge. Energy should be in Kcal/mol and forces in kcal/mol/angstrons.
Acceptable Values: "mopac" or "orca" or "custom"
Default Value: required
qmExecPath
Description: Path to the QM code executable.
Acceptable Values: path to executable
Default Value: required
qmSecProc
Description: Indicates a secondary executable that NAMD will call AFTER each QM software execution for each QM group. The executable is called with two arguments: the complete path and name of the input file used for each QM software execution; and the simulation step. This option can be used for an extra-processing at every step, e.g., for saving all QM outputs every step.
Acceptable Values: path to executable
qmPrepProc
Description: Indicates an executable that must be called BEFORE the FIRST QM software execution of each QM group. The executable is called with one argument: the complete path and name of the input file used for each QM software execution. This can be used to setup a charge distribution for a molecule of interest, for example.
Acceptable Values: path to executable
qmChargeMode
Description: Charge calculation mode expected from the QM software. This indicates if charges should be read from the QM software and updated at every step, or if the original force field atom charges should be used. In case you are using ORCA, two charge options are allowed, Mulliken or CHELPG. We must know the kind of charge requested by the user so that the proper format is expected, read and processed. NONE: No charges are read from the QM software output and the original force field charges are preserved. MULLIKEN: This is the only other option for MOPAC and one possibility for ORCA. In case you are using the custom QM software interface, choose this option in order to use the charges calculated in the custom QM software, irrespective of the actual theory used for that calculation. CHELPG: This is a second possibility for ORCA.
Acceptable Values: "none" or "mulliken" or "chelpg"
Default Value: mulliken
qmOutStride
Description: Frequency of QM charge output. A dedicated DCD file will be creatd to store the charge data for all QM atoms in the system. This independent frequency allows the user to store whole-system data at a larger stride to save time and space.
Acceptable Values: integer
Default Value: 0 (not saving)
qmPositionOutStride
Description: Frequency of QM ONLY position output. A dedicated DCD file will be creatd to store the position data for all QM atoms in the system. This independent frequency allows the user to store whole-system data at a larger stride to save time and space.
Acceptable Values: integer
Default Value: 0 (not saving)
Tutorial
A basic tutorial can be found here, but an initial explanation accompanying a simple example are available below.
Tutorial data can be downloaded for the classical simulations (~174 MB) and for the quantum mechanical simulations (~490 MB).
Basically, in order to set up a system for hybrid QM/MM simulations, one simple modification must be made to the input PDB file: the atoms which will be treated quantum mechanically must be identified by changing the value in their "beta" (or "occupancy") column to a positive integer. The chosen column must be identified in the configuration file using the "qmColumn" keyword (the column will be referred to as the "QM Column"). Different independent QM regions should be identified in the same PDB file by using different identifiers in the QM Column, that is, all atoms in QM region "1" should have the number one (1) in their QM Column fields, all atoms in QM region "2" should have the number two (2) in their QM Column fields, and so forth.
One extra atom identifier is needed when only part of a molecule is to be treated by quantum mechanics. If a covalent bond has one atom in the MM region and another in the QM region, both should have a non-zero value in their Bond Column (the column not used as QM Column, either "beta" or "occupancy"). In this case, the Bond Column must also be identified with the keyword "qmBondColumn" in the configuration file, otherwise its data will be ignored and no QM/MM bonds will be identified. An important consideration when deciding which bond(s) will separate the QM from the MM region is the distribution of charge in across the molecule. See above for the Charge Group section.
In Example1 you will find a small system and a TCL script that prepares it for QM/MM simulations. The included TCL script can automatically detect and prepare QM/MM bonds in case the the QM region is defined by simple selections of entire residues. The script is capable of handling protein and nucleic acid sequences, and can serve as a template for more complex systems.
After applying the QM region selection to the system PDB file, a selection of keywords must be added to NAMD's configuration file in order to activate QM/MM and pass the proper data to the chosen QC software. Once more, in Example1 you will find a minimal configuration file for a simulation with one QM region and a standard selection of parameters.
An advanced tutorial can be found here, and example outputs can be downloaded here (~1.2 GB). The advanced tutorial covers the use of QM/MM in the calculation of the free energy profile of a chemical reaction. We combine a String Method optimization with eABF calculation, both present in NAMD through the colvars module, and use QM/MM to simulate a decarboxylation reaction. The system is prepared using VMD, and all preparation and analysis of the enhanced sampling and free energy callculations are done in a Jupyter notebook.NAMD QM/MM Main Features
Integration to ORCA
Integration to MOPAC
Flexible and easy integration to other software using wrapper scripts
Multiple independent QM regions processed in parallel
Support & Bug Report
The first version of NAMD QM/MM is available in NAMD 2.12.
Attention, this is the first public release of a NAMD with QM/MM support. Even though the software was exhaustively tested it might still contain problems, please use caution.
If you find any problems using the QM/MM implementation in NAMD please contact us by e-mailing one of its main developers: melomcr@gmail.com, rcbernardi@ks.uiu.edu, or trudack@ks.uiuc.edu.
Any feedback is appreciated.
Training & Workshops
Applications are now open for our first NAMD QM/MM workshop, which will take place in Urbana in the Spring of 2018, from April 5th to 7th. You can check our upcoming workshops where your can learn more about Computational Biophysics here.
Collaborators
Our center focus on the development of computational biology methods and software, particularly molecular dynamics simulations tools. For quantum chemistry aplications we rely on outstanding collaborators, who assist us in the development of the QM/MM tools.
Development
NAMD QM/MM is in constant development by a team of investigators in Professor Klaus Schulten and Professor Zaida Luthey-Schulten groups at University of Illinois. The QM/MM interface in NAMD is mainly developed by:
Pseudo-Code
Click here to find a pseudo-code for the QM/MM interface.
Scientific Successes
Setting up the Genetic Code. Investigating the GluRS:tRNA:Glu-AMP complex, the aminoacylation reaction mechanism was studied to distinguish between four possible mechanisms. Uniting Network Analysis results for the full complex with the first combination of the string method, parallel eABF, and QM/MM simulations, provides an unique view for essential steps in establishing the genetic code. read more
Photochemistry in Flavoproteins. Flavin derivates are highly sensitive to photodegradation by sunlight. Dodecin, a dodecameric protein in archaea bacteria, is capable of efficient excited state quenching in flavin derivates, preventing photodegradation. Using NAMD and Gaussian for QM/MM simulations, the geometry of the protein's active site has been refined in order to calculate the UV/Vis spectrum of flavins in the protein environment. Subsequent high-level ab-initio excited states calculations led to a hypothetical mechanism of the quenching process. Visualization of important molecular orbitals was also achieved with the novel plotting capabilities inside VMD. read more
Loading orbitals from NAMD QM/MM calculations in VMD
To extend the analysis capabilities for the QM/MM suite and its supported QM packages, we implemented a molfile plugin that is able to load the orbital information from both ORCA and MOPAC. Learn how to visualize molecular orbitals using VMD.
Making hybrid QM/MM simulations easy, our next goal
Biomolecular force fields fail to address chemical reactions and other complex quantum effects near, e.g., reaction centers. Treating such parts of the molecule via quantum mechanics (QM), while the majority of simulation remains classical, allows the accurate representation of quantum effects in relatively large systems, such as enzymes. While many levels of quantum theory are available, QM/MM simulation requires the exchange of only positions, charges, and forces, enabling a common NAMD interface serving multiple QM engines.
In NAMD, a QM/MM interface to both ORCA and MOPAC has been developed. QwikMD works to prepare, with very simple steps, all the necessary files to perform these advanced QM/MM simulations.
QwikMD can now be employed to perform QM/MM simulations.


