Protein Folding Spotlights
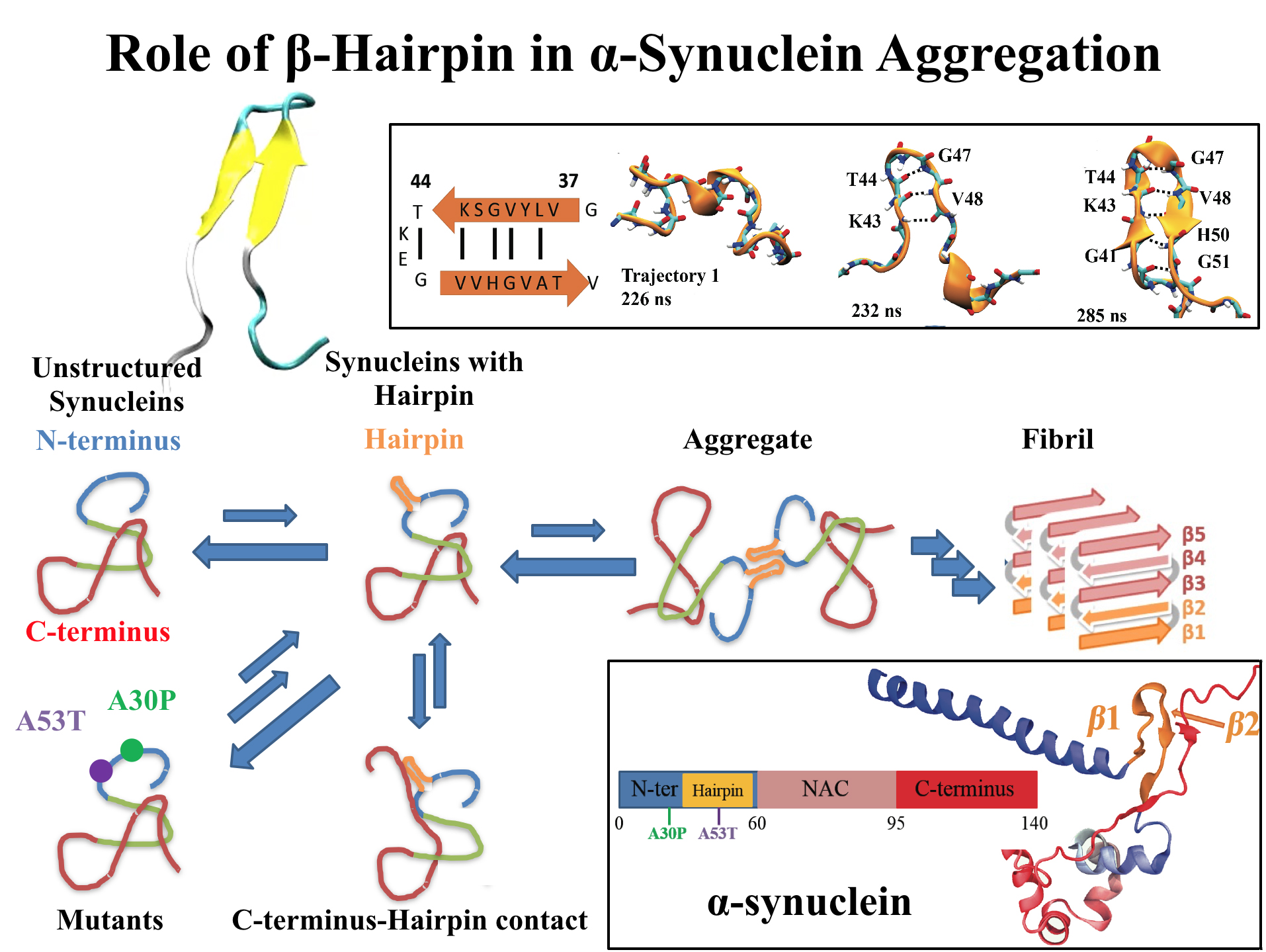
image size:
768.0KB
made with VMD
For centuries, millions of people around the globe have been troubled with a movement disorder that usually starts with a tremor in one hand. The disorder, later known as Parkinson's disease, affects commonly older individuals and disrupts patient's movement, sleep and speech from the brain. There is currently no cure for the disease. Key to the disease, progressively occurring in patient's brain, is the loss of neuron cells due to aggregation of a small protein named α-synuclein. Extensive studies have been carried out, yet the function of the protein remains a mystery. It is amazing that aggregation of such small proteins eventually leads to neuronal cell death and generates tremendous difficulties in peoples' life. In a recent report, a team of computational scientists attributed the cause of α-synuclein aggregation to a hairpin structure involving just a small region (amino acids 38-53) in the middle of the protein. With extensive simulations (over 180 μs in total), the researchers revealed that a short fragment encompassing region 38-53, exhibiting a high probability of forming a β-hairpin structure, is a key region during α-synuclein aggregation. Moreover, the researchers predicted a mutation that impedes β-hairpin formation, thereby retarding α-synuclein aggregation. The discoveries, made possible through the software NAMD and VMD, are expected to shed light on the mechanism underlying Parkinson's disease and to inspire the design of drugs. More on our α-synuclein website.
While pressure can help in cooking your favorite meat for dinner, pressure is also helping scientists to study how proteins, a key ingredient in any meal, loose and regain their proper shape. Proteins are key building blocks for any life form on earth, making the many machines that drive living cells. For any protein to do its job correctly, it has to first settle into the proper shape, the so-called native state. The process, referred to as protein folding, is still a mystery (see July 2012, Nov 2009 and May 2008 highlights). A general consensus is that the mystery can be solved only through a combination of experimental observation and computer simulation. In two recent reports (1 and 2), a team of experimental and computational scientists have used high pressure to force a protein to loose its proper shape, similar to what happens in a pressure cooker. After the high pressure is released in the experiment, the protein regains its proper shape, apparently by following two folding pathways, one fast and one slow. Using the molecular dynamics simulation program NAMD, as well as a special purpose supercomputer, Anton, the researchers were able to identify these two pathways and to follow every single folding step of the protein at an unprecedented precision. The studies greatly improve scientific understanding at the molecular-level of how proteins respond to pressure changes and, while not giving delicious recipes for pressure cooking in the kitchen, they are likely to suggest how to use pressure to dissolve toxic proteins that arise in disease, such as in Alzheimer's disease. More on our protein folding website.
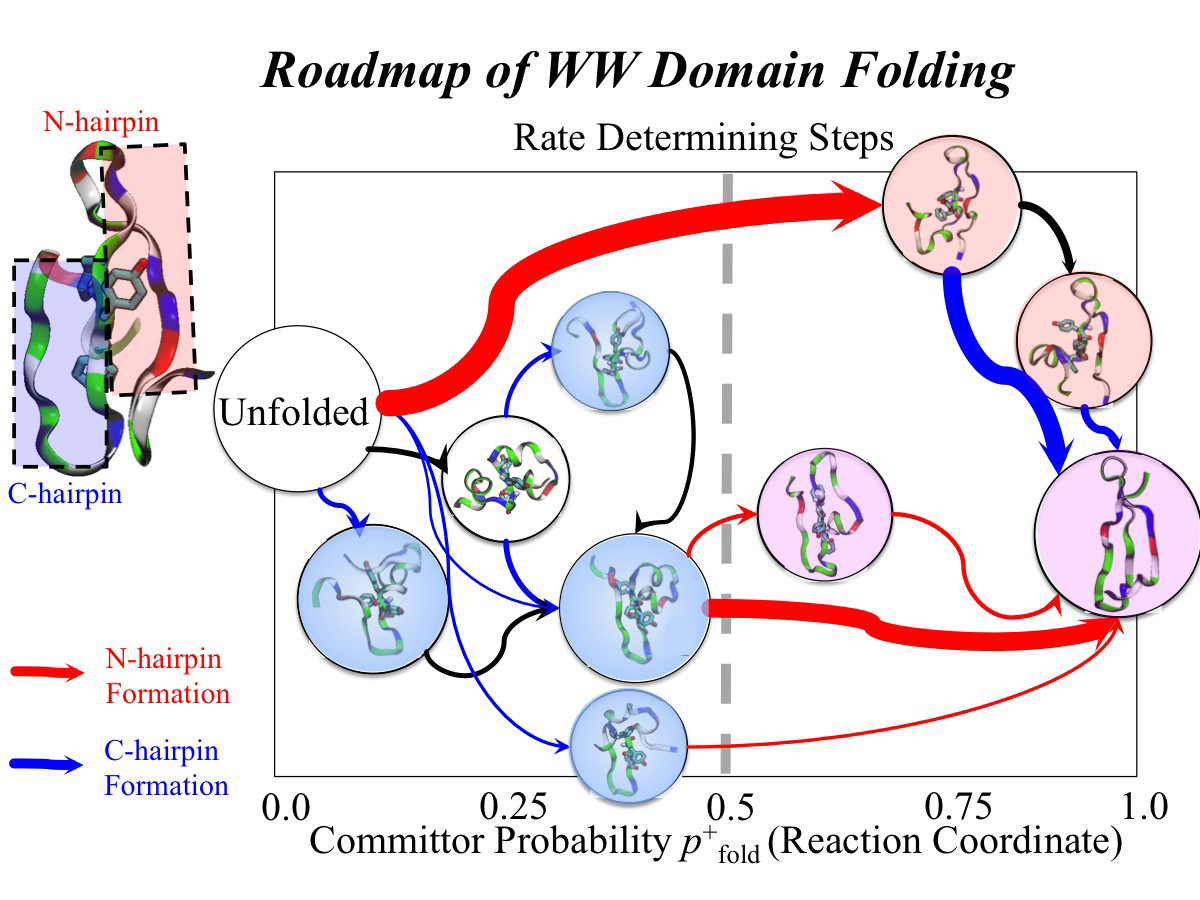
image size:
507.8KB
made with VMD
Every protein in a living cell needs to assume a proper structure in order to fulfill its biological function. The proteins that fails to do so cause malfunction of cells, often leading to disastrous diseases. Thus, how a protein folds into its native structure is one of the central biological processes that need to be understood. The modern view of protein folding suggests that a folding process is governed by structural transitions among a wide spectrum of structures that a protein could access; all of these possible structures and associated transitions constitute a complex roadmap for protein to follow, very much like the one commonly used to navigate a car to its destination through the best route. Obtaining comprehensive and accurate roadmaps for protein folding are essential for the characterization of correct folding routes. In theory, such a roadmap could be explored through computational simulations using an accurate model including every atomistic detail; in practice, the structural complexity of proteins turns the exploration of its roadmap into a daunting computational task. As reported recently, researchers have demonstrated an efficient way to explore roadmaps for protein folding by utilizing a hybrid model which combines atomic models for the protein part with a fast simplified model for the surrounding water part in order to achieve both accuracy and efficiency. By investigating folding mechanisms of two proteins, the researchers showed that their approach is able to generate folding roadmaps as accurate as those obtained with complete atomic models while only taking days to finish the task, much faster than the complete atomic models that usually need months. For details, please see our hybrid-resolution model website.
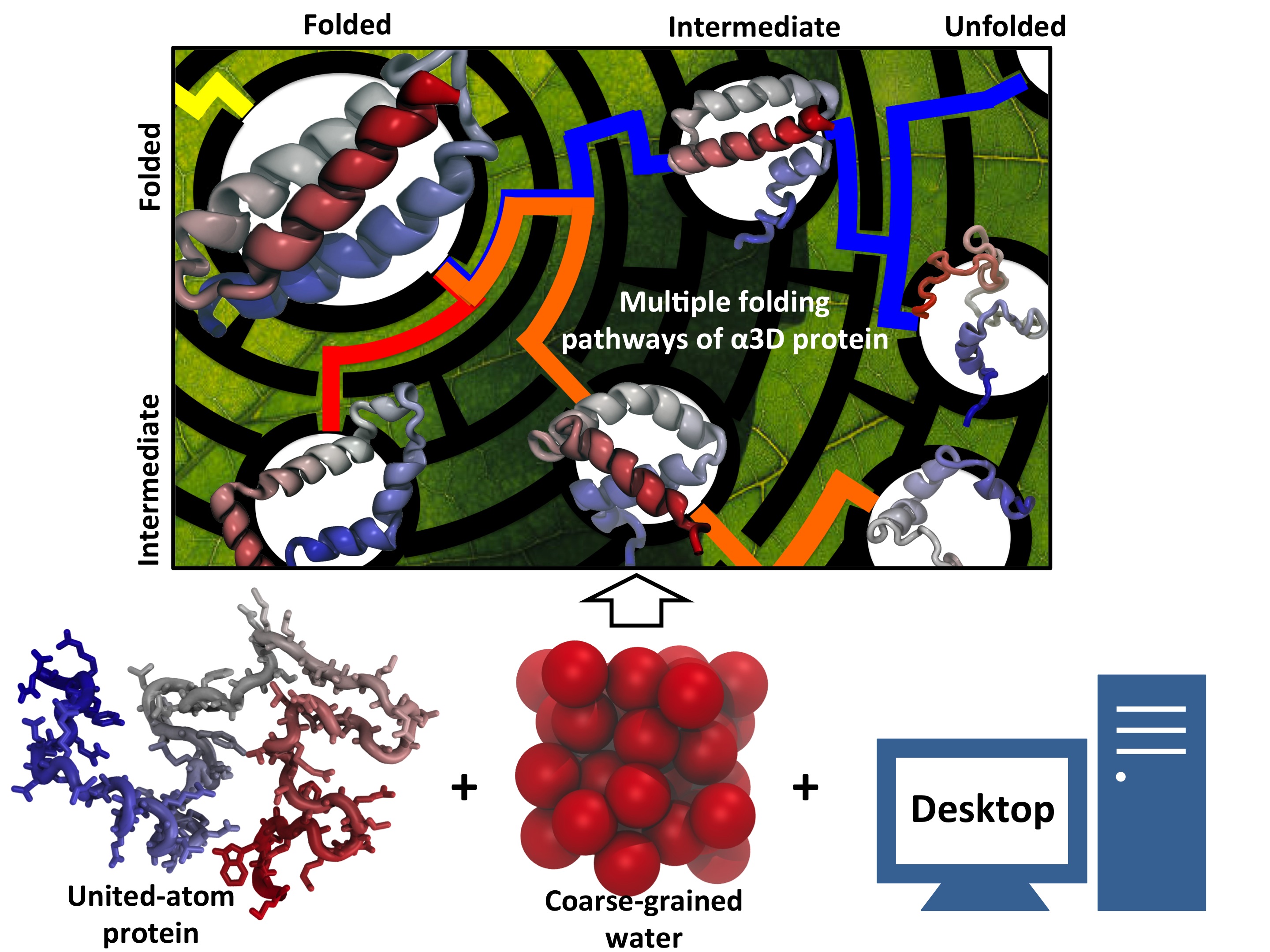
Every living cell relies on proteins to carry out its functional tasks; every protein needs to assume a proper shape in order to be operational for these tasks. How a protein, composed of a particular sequence of amino acids, could find its way to a proper shape is a fundamental, yet mysterious biological process. Researchers have sought to unravel atomistic details of protein folding processes through computer simulations, but modeling such processes is computationally demanding. It was only recently that some researchers have been able to observe in some case how proteins fold, but needed for the purpose the fastest computers available today. One of these computers is Anton, the expensive special-purpose supercomputer available essentially only to a single research group. Is there an affordable way to simulate protein folding? One solution could be coarse-grained methods. These methods save tremendous computational effort by replacing computational models that include all atomistic detail. However, the simplified models need to include a sufficiently accurate description of proteins for modeling folding processes. As reported recently, researchers have overcome the challenge by combining atomistic and coarse-grained descriptions. The new method is fast enough to follow movements of proteins long enough to see them fold, while requiring only readily available computer powers. The new method allowed researchers to analyze complete folding events for seven proteins, including a protein, called α3D (see movie, 11.5 M), that is one of the largest proteins ever folded computationally. More on our hybrid-resolution model website.
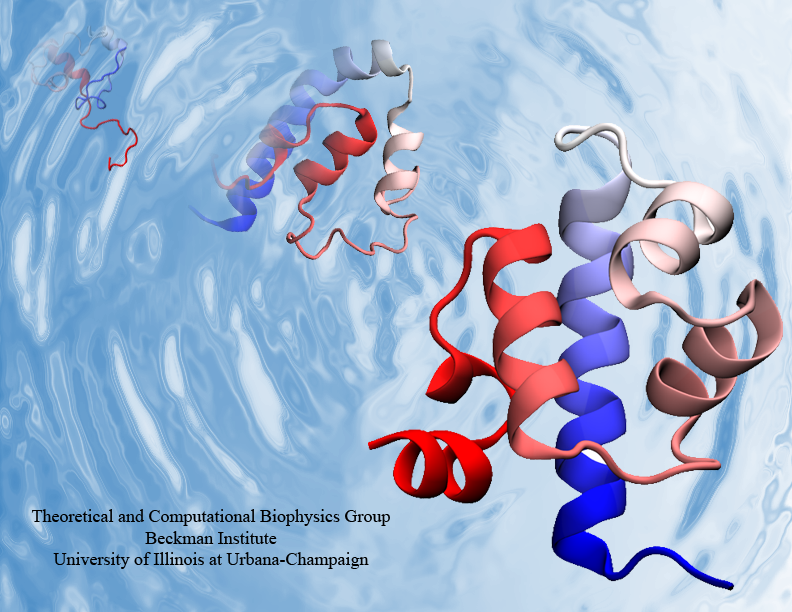
Proteins are the biological workhorses in living cells. For example, they respond to external signals arriving at the cell surface or transport cargo, much larger than themselves, from one place to another in the cell. However, before a protein can carry out his job, it must first assume the proper shape. Proteins are long polymers of twenty different amino acids linked in a linear sequence; the latter is particular for each protein. It is still a mystery how a protein folds into the proper shape based on its sequence. Scientists hope that one day they can "watch" this folding process for any given protein. The dream has been realized, at least partially, through the use of computer simulation. After tackling the protein-folding problem already computationally for two small proteins (see May 2008 highlight and Nov 2009 highlight), researchers have now successfully visualized the complete folding process of a relatively large protein, the so-called λ-repessor (see movie, 7.8 MB). In fact, it is one of the largest proteins folded to date using a computer. As reported recently, simulations carried out with the program NAMD, as well as simulations carried out on a special purpose supercomputer, Anton, achieved to follow λ-repessor's folding movement for more than 0.0001 seconds, long enough to observe the protein assume its proper shape. More information is available on our protein folding website.


