Editorial Series
Overview of the Midwest Computational Biomolecular Modeling Symposium
A whole cell.
By Lisa Pollack
November 2015"We can only see a short distance ahead, but we can see plenty there that needs to be done." -Alan Turing
Note: On September 15-16, 2015, the NIH Center for Macromolecular Modeling and Bioinformatics held a Symposium where 14 scientists from around the Midwest (see schedule of speakers.) gathered at the Beckman Institute in Urbana, Illinois to brainstorm on the future of biomolecular modeling. The following article summarizes some of the key points delivered by the speakers, and some reflections about the messages of the Symposium.
One of the hats a research scientist must wear is that of "soothsayer." Whether it be for writing grants, seeking collaborators, or choosing a topic of study, the future is seldom far away from the consciousness. In September, the NIH Center for Macromolecular Modeling and Bioinformatics decided to examine the future during their Midwest Computational Biomolecular Modeling Symposium, which it organized at the Beckman Institute in Urbana. While looking ahead is valuable on many levels, the Center had an ulterior motive as it gathered modelers from around the Midwest to speak at the symposium. In 2016 the Center goes up for renewal and as a result must propose what direction to take its tools.
The Center is specifically funded to bring new tools to the biomedical research community. For many centers, this could mean a new instrument to use in an experimental lab. However, for the Center in Urbana, the tools it offers to others are software tools. The task of the Center is to make the software foolproof for the broader user community, and then disseminate it widely. To be clear, the NIH will only fund tool development, but another part of the equation is the research that will fuel the tool (or methodological) development. While the Urbana Center has to propose what new tools to debut during the period 2017-2022, it also must propose driving research projects that will accompany the software development. For PI of the Center Klaus Schulten, this essentially means writing two renewal proposals–one on tool development, and one on research. In regards to the latter, Schulten must make a case that his tool development is science driven, and that there are projects out there the Center can work on that drive the technology into the future. However, while both aspects of the renewal writing are important, the Symposium mainly addressed the methodological aspect of the Center's mission, i.e., its tool development.
To understand why the Center held a two-day symposium on biomolecular modeling, Schulten revealed his secret for predicting the future: hard work and lots of thinking. He is the kind of scientist that prefers to ruminate before he writes his grant proposals or renewals. However, he clearly acknowledges that other people might like to work out their future plans as they write. When asked if there was a strategy to crack the opacity of the future, Schulten answered, in his characteristic way, with a joke: "It's very difficult to predict something, but particularly the future," he mused. Regardless of how difficult it is to look ahead, ten renowned modelers from the Midwest joined Schulten and three of the Center co-PIs, and all agreed to come together to offer their vision of biomolecular modeling into the year 2022.
Opening Remarks from Klaus Schulten
The Symposium began with Klaus Schulten jump-starting the day. He acknowledged that this was a very unusual workshop, for speakers were asked to forego talking about their past accomplishments and instead look at the next seven years. The upshot of the first half of his talk is succinctly captured in a slide that set the tone for the entire symposium. As can be seen in this slide, connecting the two areas that the Center will focus on beyond 2017, its tools and its driving research topics, is a bridge composed of "Challenge Issues to Address." It took months of intense discussion between Center members to settle on the "challenges" in the slide, an endeavor Schulten called "a labor of love." Other speakers picked up on the bridge motif and would use it in various ways to describe their own visions of the future.

Slide from Klaus Schulten's Talk
Leading up to the "bridge" slide Schulten gave a brief overview of the Center tools and the accompanying driving biomedical research areas. He talked about the tools the Center currently develops, mainly NAMD, VMD, and MDFF. Schulten pointed out that with continual development of NAMD, starting in 1990, systems progressed from small protein in vacuum to larger and larger proteins, and then to entire assemblies of proteins in a membrane. Schulten pointed out that this increase in system size brings the researcher closer and closer to simulating the smallest known living entity, a cell. Schulten noted that developers are currently working on remote visualization with VMD and reiterated how popular this is, for it gives the user the opportunity to sit with a laptop in their living room and have the power of a supercomputer undergirding their analyses.
While tool development is one issue the Center needs to tackle, Schulten briefly discussed the driving science areas that will accompany the grant renewal. These topics are listed on the right-hand side of the bridge slide. A key area he emphasized is whole-cell modeling. One of the Center co-PIs, Zan Luthey-Schulten already has a tool for simulating an entire cell or colonies of cells (see below). Schulten said he looks forward to combining the top-down approach of Luthey-Schulten with a bottom-up approach that necessarily takes into account the molecular level of an organism. A long-range goal is to combine these two approaches some day.
The heart of Schulten's talk came when he focused on "Challenge Issues to Address." For example, with the simulations growing larger, analysis is becoming much more tricky. In "advanced analysis," the Center envisions being able to change things as a simulation is running, such as forces or other conditions. Another aspect of the challenges is improving NAMD features and NAMD performance. Some features Center members have identified for refinement include constant pH, large systems, and polarizable force fields. And then there is the long cherished goal of increasing the time scale of a molecular dynamics simulation, with Schulten expressing interest in overcoming that "speed bump," as he phrased it.
While Schulten acknowledged the many challenges the Center hopes to tackle in the years 2017-2022, he also talked about some of the promising technologies that will provide the Center with new solutions and new opportunities going forward. First off, more powerful supercomputers are now a priority for the U.S. government (see for example this editorial about the National Strategic Computing Initiative) and researchers can expect between a ten- and one-hundred-fold increase in computing might within the next seven years. And on the science side, all sorts of advancements are happening now that the Center should take advantage of. Schulten pointed to the revolution in electron microscopy, and also that it is now possible to resolve the contents of an entire cell experimentally. Free-electron lasers are making it possible to get once-tricky structures. And, finally, structural biology is going to larger and larger structures, as Schulten noted from looking at the literature. While there are these many opportunities for the Center to utilize going forward, Schulten mentioned that funding is one issue everyone will have to tackle, and will present a definite challenge.
After Schulten's talk, there was a heated debate about large-scale modeling. Attendee and speaker Marcos Sotomayor proposed a question to Schulten directly about the utility of whole-cell simulations: "But what will you learn?" Sotomayor said this is the kind of question he sees from experimentalists all the time (especially when they interrogate modelers). Schulten countered by saying he doesn't know precisely at the outset, but added, "Why do you climb a mountain? Because it's there." Schulten finished off by concluding: "That's very much how science goes, and once you climb it you realize new things."
Sharon Hammes-Schiffer and Jingzhi Pu and QM/MM
While the NIH Center for Macromolecular Modeling and Bioinformatics may be known for molecular dynamics, the center invited two speakers who look more at the quantum world - Sharon Hammes-Schiffer, from the Department of Chemistry in Urbana-Champaign, and Jingzhi Pu, IUPUI, Chemistry. Both speakers talked about the future of the method called QM/MM (Quantum Mechanics/Molecular Mechanics).
While Klaus Schulten had shown a slide of a bridge connecting research to tools in the opening talk, Hammes-Schiffer made reference to Schulten's slide and started off saying she envisioned more of a "raft" metaphor to describe the future of QM/MM. "I give you a raft that you paddle along. You're not going to get there as fast as on the bridge, but eventually you'll get there, maybe not by 2022," Hammes-Schiffer talked about how she is interested in "Chemistry," i.e. bonds breaking and forming. She reiterated that our bodies work by doing chemistry, which is a major focus of TCBG as well, namely, to understand the function of biomolecules at the level of chemical detail.
One of the major areas Hammes-Schiffer described was QM/MM that allows for conformational sampling, and she talked about what might be possible for this method looking far into the future. She noted that this could be relevant to TCBG because Schulten had mentioned his interest in bioenergetics systems in the opening talk, so Hammes-Schiffer said she realized Schulten was interested in electrons, protons, and proton-coupled electron transfer, which are important in photo-initiated processes.
Hammes-Schiffer reviewed some of the current methods for QM/MM that allow conformational sampling (like empirical valence bond fitting and semi-empirical methods), but then talked in detail about combining umbrella sampling with a finite temperature string method. While there is currently a CHARMM-QChem interface to do these calculations, Hammes-Schiffer believes NAMD could adopt an interface for this type of approach without much effort by the developers.
There was a lively debate after Hammes-Schiffer's talk between Center faculty, where Alek Aksimentiev asked if one even needs quantum mechanics to describe a cell, and Zan Luthey-Schulten responded by saying that the rate constants Hammes-Schiffer calculates are already making a major contribution to cell biology. Luthey-Schulten in fact does whole-cell simulations (see below) and is always on the hunt for rate constants. The lively debate ended with discussions of what methods might be most suitable for different systems, and Hammes-Schiffer reiterated that "the method you use depends on the questions you want to ask."
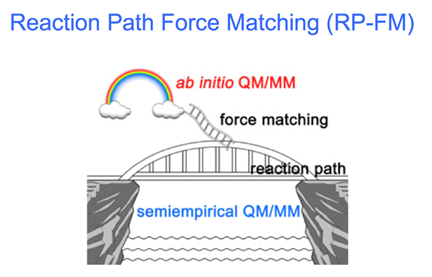
Jingzhi Pu Slide on Reaction Path Force Matching
"Back to the Future of QM/MM" is the slide that greeted attendees of the talk by Jingzhi Pu, making reference to the 1980s movie and introducing a bit of humor. Acknowledging that the 2013 Nobel Prize in Chemistry was exciting and validating for those in the QM/MM field, Pu posed the question "Is this bad news? Is the party over for young researchers?" He was trying to make the point that if one wants to continue working on development of QM/MM, one has to think hard about the future opportunities. He then showed a curve of QM/MM citations over the past twenty years and all attendees could clearly see that these citations have grown tremendously in two decades. Pointing out that many of the recent papers are applications only, Pu concluded that in the future, one should make QM/MM more accessible to end users, as well as user friendly. Other features he thinks would be desirable to users included having a graphical interface to define selections for QM regions, or coupling VMD's force feedback to QM/MM potentials so a user can move the bond around to "feel" how good the reaction mechanism is. John Stone, lead developer of VMD, pointed out that Todd Martinez has a prototype of interactive QM/MM already, using VMD and TeraChem.
What direction is Pu taking in his development of QM/MM? Should the future be ab initio or semi-empirical? The answers are conveniently explained in a slide, which is in keeping with the bridge and raft metaphors.
After explaining the many problems inherent in trying to accelerate ab initio QM/MM, Pu said the approach he is taking is to increase accuracy of semi-empirical methods. His incarnation is called Reaction Path Force Matching, and the slide shows a bridge that represents a free energy reaction path, which can realistically be achieved from computation, and a rainbow for an idealized path that is out of reach of current computing power. The ladder thus bridges the gap between ideal and reality. As Pu pointed out during the Q&A that followed, if there's no drastic change in computing technology, we have to live with semi empirical method, but with the caveat, chimed in Hammes-Schiffer, that it has to be re-parameterized for each system.
Diwakar Shukla
The next talk was a vastly different topic, but one dear to the hearts of TCBG, on what is the future of molecular dynamics. University of Illinois professor Diwaklar Shukla started out his talk by saying he would like to look at "the future" by rethinking the question to read: How can we transform molecular dynamics into an enabling tool for disruptive science? Shukla said he likes that formulation of the question because it offers a more broad perspective.In 1990 Martin Karplus and Gregory Petsko wrote a paper in Nature where they listed the limitations of molecular dynamics as 1) approximations of potential energy functions and 2) length of simulations. Shukla went on to say that, twenty-five years later, we still have the exact same challenges. But Shukla added that "accuracy" is an additional challenge he sees for the field. His examples to illustrate this accuracy challenge were population states of kinases and protein folding (getting the correct kinetics). Klaus Schulten wanted to know what actually constituted the "right kinetics?" Shukla said he sees this as a dimension of the accuracy challenge, for the community needs to come up with standards to compare against when looking at kinetics.
The final challenge Shukla mentioned related to what to do with so many simulations of biological systems; he believes the next step is to synthesize that information into something bigger than the individual studies. He used as an analogy, to drive home his point, the study of individual planets versus the entire picture of celestial dynamics.
Shukla closed out his talk by pointing to future directions. He talked about researchers doing protein in a box, versus those doing whole cell simulations, but wondered if there is an intermediate level of resolution that could actually give more information. As far as application areas, Shukla is wholeheartedly behind expanding molecular simulation to non-traditional areas. One area where he sees potential is the water, food, energy nexus; that is, all three areas are individually connected (one needs water to grow food, one needs energy to purify water, one needs food for energy, etc.) As an example of this, Shukla talked about his work looking at plants at the molecular level, that is, modeling plants in silico. One thing very exiting to him after joining this field is, compared to his work in medicine, experimentalists can generate results in four months instead of years.
Dong Xu
In keeping with the theme of bridges, Dong Xu (U. Missouri) titled his talk "Building a Bridge between Molecular Dynamics Simulation and Protein Structure Prediction." Xu was a graduate student in Schulten's group in the 1990s and now he is the director of the Digital Biology Laboratory, whose tag line reads, "Explore biology and medicine computationally." Although he has many research areas in the Laboratory, he chose to talk about the ways protein structure prediction and molecular dynamics could benefit each other. Xu is an architect behind the structure prediction program MUFOLD, one of the many tools from the Digital Biology Laboratory.

Dong Xu, from Missouri, who talked about structure prediction.
Xu started his talk with some intriguing statistics to reinforce the idea that protein structure prediction is a valuable tool looking forward. As a protein's structure is a key element needed to elucidate function, there are only just over 100,000 structures of proteins in the central repository for such structures, the PDB (Protein Data Bank). But, with the current cost of genome sequencing growing smaller, there are over 50 million sequences now available. This is a ripe opportunity for structure prediction tools, to capitalize on the waiting market of sequence data.
As for the future of structure prediction, Xu identified many areas he sees the field heading toward: cloud computing, more easy-to-use tools (eg. FoldIT), and ability to solve the structure with fewer data (like the program Rosetta does currently with NMR data). But the heart of Xu's talk elaborated on the theme he chose to focus on: how structure prediction and molecular dynamics could mutually benefit each other. Xu believes molecular dynamics could be used for model refinement, model selection, and function studies. For example, he pointed out that dynamics can often provide a better probe of model stability than static scoring functions. Another useful suggestion he had was to integrate a structure prediction tool into VMD and NAMD. This would benefit VMD and NAMD, by giving them more functionality, but also the world of structure prediction could be introduced to the large base of VMD and NAMD users. A further suggestion was to apply techniques to handle big data to molecular dynamics, turning "big data into smart data." He gave an example of using deep learning to identify the difference between two states of a system when finding the answer is not so clear cut to do by hand.
Another area of overlap Xu mentioned was in the task of molecular replacement (i.e. the phase problem for X-ray diffraction data). So far, using low-quality models does not seem to work for molecular replacement, but by using molecular dynamics simulations, Xu sees potential to make the low-quality model usable for final structure determination. In the Q&A after the talk, there was much enthusiasm from Klaus Schulten about the topic of molecular replacement. Schulten also expressed interest in collaborating with Xu in one of the TCBG's newest products, xMDFF. (This recent edition to the Center tools, xMDFF, makes it possible to refine the structures of low-resolution X-ray diffraction data.) Schulten mentioned a feature in xMDFF that allows interactive refinement, or a way to provide immediate feedback about quality of fit. This interactive refinement is especially useful for targeting local places in the density where the fit is not so great, and which may not be reflected in the overall score indicating quality of fit. Schulten thanked his former student profusely for many good suggestions.
Marcos Sotomayor
The moderator of the symposium introduced the next speaker, Marcos Sotomayor, as the only experimentalist on the schedule. Sotomayor elaborated humourously by saying, "people from the computational labs say this is the experimental guy and people from experimental labs say this is the computational guy." To be clear, Sotomayor got his PhD in the Schulten group in the mid-2000s, but then went on to study crystallography in his postdoctoral position. Now Sotomayor runs a laboratory at Ohio State University that combines experimental techniques with computation. So it seemed only fitting that Sotomayor said he wanted to deliver his talk today from the perspective of a user. He promised the room many suggestions from his group about ways to make the software more user friendly.

Marcos Sotomayor, a professor at Ohio State University
Before he launched into suggestions, Sotomayor talked about the biology he does in the lab, studying specific proteins related to mechanotransduction and cellular adhesion, namely from the family of proteins called "cadherins." For example, two cadherin proteins (protocadherin-15 and cadherin-23) form a filament that pulls open transduction channels in the hair cells of the inner ear. The filaments are also called "tip links." A concrete example Sotomayor gave to illustrate relevance of cadherins in daily life involved going to a rock concert and standing next to a speaker, which causes huge amounts of force on the filaments. If the loud music ruptures the tip links, the rock concert fan will be deaf until the tips heal. Sotomayor said a goal of his, computationally, is to simulate the whole transduction apparatus (i.e. the transmembrane domain, the transmembrane channel and roughly 40 extracellular domains ). This long-range goal will involve 5-10 million atoms. Another project he has on tap is to simulate cellular adhesion linking two classical cadherins, which could amount to 10-20 million atoms.
Sotomayor identified three areas he thought would make a difference to an everyday NAMD and VMD user: speed, data handling, and structure building. He said he garnered feedback from all his students and postdocs, who regularly use the software. Speed was just discussed in the Diwakar Shukla talk, but Sotomayor pointed out that signaling usually is on the millisecond time scale and this would be great to reach in a simulation. With systems growing larger and larger over the years, handling these big data, such as in the form of trajectory files, could be made simpler in VMD. And finally, structure building might take some cues from the crystallography software that his students find so easy to use.
At the heart of Sotomayor's talk was what he thinks are some coming trends in biology. He talked about the new equipment that allows people to monitor whole embryos or organs as they are allowed to evolve. He also mentioned Oculus Rift and nano-reactors. He then spent a good deal of time discussing how excited he sees people over possibilities with gene editing. Sotomayor said it is difficult to simulate RNA (especially with current force fields) but that a revolution is happening in this field. He said it is good to ponder: what does molecular dynamics need to do to simulate this successfully?
He closed by hinting that perhaps people want to even look beyond whole-cell simulations, to colonies of cells, such as would be found in developing embryos. One would not only need to determine the level of detail needed for each cell, but also some level of understanding about how the cells connect to each other. He said technology is now providing very detailed views of how cells together form an organ or an organism, but there is no computational tool that allows us to simulate and predict that right now.
Andrew Ferguson
Speaker Andrew Ferguson, representing the Material's Science Department at the University of Illinois, gave a talk titled, "The Role of Machine Learning in Biophysical Assembly and Folding." Ferguson identified a theme he witnessed from the previous talks, namely, integrating experimental data with molecular code, and said he would definitely touch on this topic throughout his talk.

Andrew Ferguson from the University of Illinois
The talk started with reference to the "data deluge" that Ferguson said society has been witnessing lately, especially notable in the last two years. The cost to sequence entire genomes has decreased markedly, providing, for example, mountains of data on this front. Ferguson also mentioned that, with the advent of faster processors and better algorithms, more simulation data is out there as well. But all these data could mean opportunities for simulation researchers, for it could allow them to interpret and integrate computation, or even help in specific ways, like the improvement of force fields. But this will require new tools, and right now more data do not necessarily mean more understanding. But Ferguson sees machine learning as one possible tool for better understanding of large data sets.
He illustrated how he is using machine learning in his own work with two examples. First, he works with an experimentalist who studies Janus Particles, and tries to make sense of the underlying mechanisms in the large data sets. He uses a method he calls "diffusion map" to see patterns in the data. The other example he gave was protein folding of an alkane chain. Here he attempted to go from a low-dimensional data set to find a multi-dimensional folding landscape. In other words, he asked, "Can we infer the geometry and topology of the folding funnel from only the evolution of a single coarse-grained observable?" With the help of Taken's Theorem, the answer was "yes."
Ferguson ended with his vision for where machine learning can really make an impact on molecular simulation: Finding underlying mechanisms, bridging the divide between experiment and computation by doing quantitative or semi-quantitative comparisons, and also perhaps guiding experiment and simulation. His final suggestion was to work to squeeze the most out of big data.
Narayana Aluru
Narayana Aluru, from the UIUC Department of Mechanical Science and Engineering, started off his talk saying his group does bio-inspired engineering, as well as takes engineering to biology. He had an opening slide that listed some of the systems he sees will be important on the horizon of this field. First, he indicated that biomolecular structures have a number of engineering uses: sensors, filtration (water desalination), separations (of gases) and molecular diodes. On the horizon he also sees synthetic biology becoming important, as it relates to bio-inspired channels, cells, and tissues. Another area he sees of growing significance is integration of nano-materials with biomolecular structures. Lastly, he noted that systems are getting so much larger that exascale computing will probably be needed, as will coarse-grained and multi-scale models.

Narayana Aluru, from Mechanical Science and Engineering at UIUC.
Aluru illustrated how his group does bio-inspired engineering with a number of examples. First, he talked about water desalination. While carbon nanotubes looked promising, Aluru said the future may be in two-dimensional membranes, such as graphene, molybdenum disulfide, and hexagonal boron nitride, nanomaterial on which he has run simulations. Another area where engineering goes hand-in-hand with biomolecules is for DNA sequencing. He talked about possible proteins that can act as pores for identifying DNA base pairs, specifically alpha-hemolysin and MscL.
An exciting field Aluru works on is cell-like constructs in synthetic biology. He talked about droplets made of water, lipids, and oil, and specifically what happens when one brings two droplets together and puts a protein like MscL in between. There is a conducting path between the two droplets. His simulations revealed electrochemical gating of MscL. But to move forward on this system, Aluru predicts simulations will have to reach larger and larger lengthscales, and probably will require exascale computing combined with coarse-grained models and experimental data. Lastly, Aluru discussed molecular diodes, which are made of source and drain and a membrane with thousands of nanopores. Again, he stressed how huge these systems are and how one probably needs to resort to coarse-grained and multi-scale models looking ahead.
Erik Luijten
Erik Luijten started off his talk asking, "Is there still room for coarse-grained models?" A professor at Northwestern University, Luijten hails from Materials Science and Engineering department. He works on systems that are traditionally much larger than atomic length, such as colloidal particles that are tens of nanometers all the way to microns, as well as all sorts of polymer physics problems. He said he hoped the examples he planned to discuss would allow everyone to see what useful techniques could be ported to biomolecular questions.

Erik Luijten, from Northwestern University.
Luijten discussed the broad picture of what he does in his own lab. First, devise new algorithms that accelerate exploration of phase space; second, give proper treatment to electrostatics, especially dielectrics; third, garner a truly mechanistic understanding of many-body phenomena. He said that what he loves about many-body physics, as well as electrostatics, is that he often encounters things that are somewhat counter-intuitive.
As an example of the kind of coarse-grained studies he does, Luijten talked about two "vignettes," as he called them. First was self assembly of nanoparticles for gene delivery, and second was the role of dielectrics in self assembly driven by electrostatics. Luijten works with experimentalists who study micelle-like objects, which are basically formed by condensing plasma DNA with oppositely charged poly-electrolytes. These objects' intended use is for gene delivery. The experimentalists came to Luijten to help with the questions: Why do these particles have these shapes? How is the DNA arranged within these particles? The experimentalists wanted quantitative insight. While Luijten was able to offer some insights, he wondered what balance can be struck between the need, on one hand, to use coarse-grained models and, on the other hand, to have something that offers better quantitative predictions. Essentially he wants to improve his coarse-grained models to be more sophisticated. Luijten stressed again that he is fascinated about this topic because it is an instance of a many-body effect that is not completely intuitive.
The second "vignette" Luijten put forward was colloids and nanoparticles. He specifically looked at the situation where they changed the solvent, that is, changed the background permittivity. Namely, he looked at the case of a high permittivity (water) and then to a low permittivity (organic solvent) and what he noticed was an asymmetry in the assembled structure. He wanted to show this dramatic result to drive home the point that dielectric effects really make a complete qualitative difference in the structure that's assembled, so ignoring them sees a bit rash.
In closing, Luijten remarked that he's very interested in coupling coarse-grained models to atomistic models in a way that yields feedback, with the goal of improving coarse-grained models. He acknowledged that current coarse-grained models are not yet good enough.
Jean-Pierre Leburton
In the talk given by Jean-Pierre Leburton, he started by promising to discuss a topic much different than the previous talks. Namely, he attempts to use solid-state nanotechnology to "interrogate molecular biology." Leburton is a Beckman Institute professor who also hails from the University of Illinois Electrical and Computer Engineering Department.
These two fields, nanotechnology and biology, were usually separate fields, but the "bionano" field now seems poised to grow, Leburton said, as the interface between the two becomes more clear. However, Leburton pointed out a major difference between biological systems and solid state nanotechnological systems: biological systems are extremely slow and unreliable, whereas solid state nanotechnology is fast and reliable. The "bridge" theme, originally presented by Klaus Schulten in Schulten's opening talk, came up again, as Leburton said his work aims to build a bridge between biology and nanotechnology, to exploit the properties of each separate component to make an overall system that runs better. As semiconductors get smaller and smaller, soon probably close to a diameter of 5 nm, this is reaching the length scales of DNA's diameter (1nm). So this is one example of a bridge, bringing DNA to nano-sized transistors, to eventually make possible portable DNA sequencing devices.

Jean-Pierre Leburton, from the Electrical and Computer Engineering Department at UIUC.
Leburton proceeded to give a history of the progress that has been made in the field of using nanotechnology to sequence DNA. This idea hinges on creating a nanotechnology "membrane" with a pore, through which one could thread DNA, and each base pair should give a unique conductance in the nanotechnology membrane. Early attempts to do this used silicon-based membranes, but then seven years ago along came graphene. This material is basically a monolayer thick, around 3 Å, and it is comparable to DNA base pair stacking distance, 3.5 Å. By combining graphene with dielectric membranes, it is possible to tune the conductivity of graphene for a better read out. However, the simulation of such systems is not trivial, and requires a combination of molecular dynamics for the biological side with quantum mechanical calculations for the electronic side. Another promising material to use as a membrane is molybdenum disulfide, since it is less hydrophobic than graphene and also is a tunable semiconductor.
While Leburton discussed detecting DNA base pairs with solid state nanopores, he also said this bionano technology looks promising in epigenetics, namely it may be possible to detect methylated DNA. One of the ways to accomplish this is use multi-layer membranes, and each layer would have a particular function, with the ultimate goal to control the motion of DNA and reduce noise.
As far as the future, Leburton sees many areas in simulation to improve upon. First, do more investigations of the functionality of the multi-layered membranes. Second, develop an approach that captures the dynamical response of the system compared to the quasi-static approach currently in use. And finally, merge the molecular dynamics with the nano-electronic calculations in a more comprehensive way.
Benoit Roux
Hailing from the University of Chicago, Benoit Roux started off his talk by noting that he was going to speak about issues related to molecular dynamics that are either unresolved or that he is in the process of resolving. He wanted to illustrate with the use of an application that he has been working on, namely the calcium pump. This membrane protein burns ATP to pump calcium one way and protons the other. His approach has been to look at the transitions between two stable states, doing two at a time, and he's been using the string method with swarms of trajectories, which allowed him to determine the mechanical pathway.

Benoit Roux, from the University of Chicago.
However, his work on the calcium pump led him to generate a list of issues he deems important for future consideration. Namely: force fields, protonation states, sampling, thermodynamics, and kinetics–hence reiterating similar issues in previous talks that also focused on problems with force fields, sampling, and kinetics. Of this list of challenges for molecular dynamics going forward, he said, in terms of the first item, that polarizable force fields look promising and, for example, the DRUDE force field is not that costly to run in calculations. This force field is now in NAMD, as well as in OpenMM and Gromacs.
Roux spent a great amount of time discussing sampling, and pointing to the few tools out there to increase sampling. He mentioned temperature Replica Exchange Molecular Dynamics, SGLD (Self Guided Langevin Dynamics), and accelerated Molecular Dynamics. To tackle this issue, he started developing a tool in his own lab about two years ago, which is based on hybrid non-equilibrium molecular dynamics Monte Carlo methods. Roux elaborated on the details of the flavor of this method that he is working on. He has a 2015 paper on this with Yunjie Chen, which covers the details he discussed at length: "Constant-pH Nonequilibrium Molecular Dynamics-Monte Carlo Simulation Method."
In the discussion afterward, talk about how to determine the kinetics, either through simulation or from experiment, led Benoit Roux to offer the following analogy about the field of molecular dynamics. He said it is a bit like sitting between two chairs. In one chair is the longing for validatation, taking a very complicated biological problem and getting correct results. In the other chair there's pressure to do predictions. "We cannot just spend the next twenty years calculating things that have been measured experimentally to show we get the right answer," Roux said. "At some point the funding agency will be running out of interest."
Urbana Center Members Zan Luthey-Schulten and Alek Aksimentiev
Although the primary goal of the symposium was to bring in outside modelers to share their views on the future of the field, Urbana Center faculty also talked about what they see is in store for themselves looking ahead. As such, there were talks by Zan Luthey-Schulten, UI School of Chemical Sciences, and Alek Akesimentiev from the UI Department of Physics.
While there was much debate back and forth during the entire symposium about the wisdom and problems of whole-cell simulation, Luthey-Schulten actually works on a piece of software that takes a top-down approach to whole-cell simulation, called Lattice Microbes. Lattice Microbes does not describe cells at the level of chemistry, such that you can see the atomic and even electronic detail of the constituent parts, but instead Lattice Microbes looks at the cell as a spatially distributed network of reactions. And to add spatial heterogeneity, diffusion is taken into account as well. In her Symposium talk, Luthey-Schulten described the basics behind Lattice Microbes, and reiterated that the starting place for her simulations comes from experimental data (tomograms, single molecule studies, and proteomics, to name a few). She also said she seeks experimental validation for her simulations, reiterating that the simulations came first. In her description of applications, she pointed out that she's learned how to correct for gene copies, as well as metabolic networks.

Zan Luthey-Schulten describing different methods for different time scales. (Molecular Dynamics, Brownian Dynamics, and the Reaction-Diffusion Master Equation)
And then she asked, what does the future hold? Luthey-Schulten thinks one interesting direction would be how to resolve sub-populations and the diversity that's among them. And, it seems, for the model organism E. coli, scientists are still discovering new reactions, whose data she can utilize. She also discussed the possibilities around the burgeoning molecular dynamics studies of the E. coli outer cell wall. And she wants to improve models for how DNA is folded. Going to a model organism like yeast, because it's eukaryotic, means taking into consideration its many compartments, a definite challenge. Lastly, she talked about the problems inherent in handling big data, which she must consult constantly for her simulations.
Another Center co-PI, Aleksei Aksimentiev talked at some length about what he sees for the future. He started of by pointing to the weaknesses of force fields, especially in regard to how charge-charge interactions are described. He used as an example the dopamine transporter. Aksimentiev's basic point was that the software tools and computer power have outpaced force field development, and he hopes this can be addressed.
Next, Aksimentiev presented his version of large-system modeling, a methodology that he calls "patchwork molecular dynamics." The upshot of patchwork MD is summarized in Aksimentiev's statement, "There is no good reason (other than convenience) to simulate big systems synchronously." This fired up Klaus Schulten, who wanted to know if one patch represented a whole protein, for he noted that the cell has many proteins interacting with each other (for example, the expression machinery). Zan Luthey-Schulten asked for more clarification on what the patches represent, and asked if Aksimentiev would break it down by cellular process. Aksimentiev answered by saying a patch could be one protein, or a conglomerate of proteins. This part of the symposium definitely produced the most lively exchanges between all the attendees, as many people debated for quite some time about how patchwork molecular dynamics might someday work.
In the end, Aksimentiev concluded by saying he would like to figure out a way to integrate genomic data (which is one of the fastest growing fields) with molecular structure. Right now, "omics" data is completely disconnected from the molecular scale.
Concluding Remarks from Urbana Center Member Emad Tajkhorshid
University of Illinois professor Emad Tajkhorshid presented the final talk of the entire Symposium, where he summed up the tentative vision going forward regarding the NIH Center for Macromolecular Modeling and Bioinformatics. He started off with a list of important considerations, such as identifying key research areas that can benefit most from computation, defining an overarching theme, and having overlap between what the center does in the future and what it currently has momentum in. He also mentioned the need to study systems that can exploit supercomputing resources.
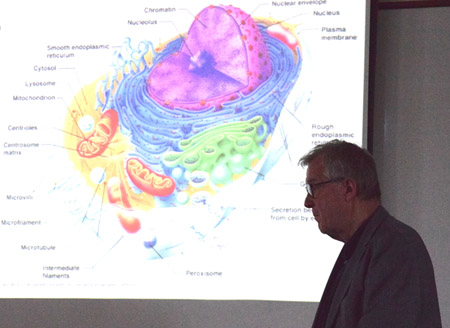
Klaus Schulten stands in front of a slide illustrating the many compartments of the eukaryotic cell.
Tajkhorshid then took some time to discuss his own research area, membrane transporters, mainly to illustrate some of the problems the Center, but more broadly also the community, will have to tackle for the future. Since membrane transporters undergo large conformational changes, one of these problems is determining the correct pathway between the two states. And on top of this, there is often the need to study other molecular events (i.e. ligand or substrate binding) as well as conformational changes. Tajkhorshid said it looks increasingly like it won't be possible to use "brute force" molecular dynamics anymore since the time scales of the systems he and many others study are so long.
Tajkhorshid then returned to talking about the vision for the Center, where he identified key areas that can benefit from the Center's computational development. These research areas include: biological systems, drug design, bioengineering applications, device design, and material science. On another note, Tajkhorshid talked about the tentative overarching vision for the Center: moving toward cellular processes. One of the key aspects to whole-cell modeling is building the structure, for how to accomplish this is not so clear. However, Tajkhorshid said another part of the overarching vision that goes hand-in-hand with structural modeling is using molecular dynamics to generate structures, which is now done routinely with the Center's tool MDFF. Another obstacle to tackle with celluar modeling is to simulate the many different time scales going on in a cell. Tajkhorshid then put up a slide of an eukaryotic cell to illustrate that complex membranes also need to be represented.
The talk concluded with a movie from Harvard (on the mitochondria) that Tajkhorshid wanted to show to illustrate the various levels of complexity inside a cell. Klaus Schulten got vary excited and said that this movie was better than looking at one protein at a time in the cell. Schulten exclaimed, "I think our job in the next decade is to turn this into real science!"
Experimentalist Weighs In
All the speakers at the Symposium, plus the majority of the audience, were computational and theory people. This begs the question: Why should experimentalists care about the future of computational modeling? Why is the Symposium important for experimentalists, too?
To answer this question, I talked to invited speaker Marcos Sotomayor immediately after the Symposium ended. He is in a unique position, as he conducts both experiment and simulation in his laboratory at Ohio State University. I asked him why experimentalists should care about the topics at the Symposium. Sotomayor replied with a favorite quote of his: "What I cannot simulate, I do not understand."
Sotomayor's main point was that it's very useful for experimentalists to have models that help guide experiments as well as explain things one cannot see with experiment. Additionally, simulation often provides predictions for experimentalists to test. For example, Sotomayor talked about forces. Recall that he examines the forces necessary to rupture the tip links of his cadherin proteins. An experiment might reveal the amount of force necessary to break a tip link, but simulation shows the underlying mechanisms involved; simulation actually takes on the role of a computational microscope, offering vistas where experiment oftimes is blind.
Another area where simulation and experiment go hand-in-hand is the feedback look. Simulation may provide information biologists can use to hone their experiments; and experiment may provide critical information when running simulations. "So I think that loop is really essential," said Sotomayor.
Experimentalists are reaching out to modelers more and more these days to collaborate. One reason for this, according to Sotomayor, is that modeling and simulation have reached a level where they can be directly compared to experiment. This is most likely because simulations attained time scales and length scales of interest to experimental biologists. And perhaps, adds Sotomayor, experimentalists are going more "nano." "So experimentalists usually like what will be useful for them, and this matching of scales I think is becoming more and more useful for both [experimentalists and modelers]."
On a side note, Sotomayor also gave his general impression of the Symposium after hearing all the talks. He was especially interested in Zan Luthey-Schulten's discussion of her whole-cell models. He notes a trend he sees in biology right now, of scientists moving towards large-scale and whole cell. But he also sees biologists going beyond that, to things involving whole tissue or an organ, or even a whole embryo. "So I think people are going in that direction and I think that would be an interesting topic to cover now," concluded Sotomayor.
Reactions from TCBG Members
The group Klaus Schulten runs, the TCBG, has been fervently brainstorming for months about what tools and methodological developments to propose for renewal of the Urbana Center. As such, group members have their finger on the pulse of modeling's bleeding edge. Did they find the Symposium helpful? Insightful? Did it confirm what they've already been contemplating intensely? Many group members offered their reactions after the event ended.
Postdoc Jodi Hadden weighed in on several fronts. Two speakers (Sharon Hammes-Schiffer and Jingzhi Pu) both suggested NAMD implement a QM/MM code, and Hadden liked the sound of this. One of her research projects focuses on cytoplasmic dynein, a motor protein, and she and other TCBG members who work on motor proteins all came to the conclusion independently (before the Symposium) that having QM/MM in NAMD would be useful for motor protein research. "It's inspiring to know that the functionality we are interested in implementing will also support the research of other laboratories with whom we do not directly collaborate," Hadden said.
Hadden also wrote an elegant paragraph on structure prediction:
The challenges of structure prediction were also covered in depth by former group member Dong Xu. Structure prediction is a particular strength of computational methods, allowing researchers to model missing pieces from experimental structures that were not resolved for one reason or another, usually because those regions are very flexible. In some cases, experimental structures are not available at all, perhaps because the protein of interest has proven too difficult to crystallize thus far, and it is entirely up to computational structure prediction tools to generate a structure in the meantime. The ability to generate complete, fully atomistic molecular structures through combining experimental data with computational predictions underlies our ability to perform bimolecular simulations to study the dynamic machinery of the living cell. In that case, as Dong Xu emphasized, it is critical that we maintain a focus on improving structure prediction, alongside our planned advances in molecular simulations. Accordingly, our group has a major core section of the planned proposal dedicated to the structure preparation step of simulation studies, including the incorporation of structure prediction tools like Rosetta into MDFF.
For postdoc Rafael Bernardi, what resonated for him was the message to make NAMD and VMD easier for new people entering the field, for he realizes that the barrier to entry is high. Bernardi really took to heart Marcos Sotomayor's talk in which Sotomayor pointed out how students find crystallographic software so easy to use for structure manipulation. "Of course NAMD and VMD are much more powerful together, but the power is useless if you cannot get new users, new people to go for this field," said Bernardi. "I think that's something that came out, and that we are already discussing in the group."
A common motif that postdoc Till Rudack noticed from all the talks was the need to "integrate." "It's not only integrating experimental results to simulations," Rudack explained. "It's also integrating different ways of simulation and simulation approaches, and combining everything together." For example, scientists may know lots about a single protein, but Rudack wants to now combine many proteins together. For Rudack, he feels like this integrating will result in giving users a more thorough "tool kit."
Reflections from Klaus Schulten

Heated debates sometimes ensued at the 2015 Symposium.
Klaus Schulten and Center members have literally been pondering the future of biomolecular modeling for many months, but all of their discussions were internal. Now, with the Midwest Computational Biomolecular Modeling Symposium, the Center literally opened the door wide for outsiders to contemplate the future. "And that is of course always very healthy," comments Schulten, "because partially they reinforce you, but partially they bring new views." Schulten called this endeavor a "reality check." Now, he says, it's back to the drawing board for Center members, who will use what they gleaned at the Symposium to rethink their renewal.
But what were the messages Schulten took away from listening to a day and a half of talks about the methodological future of the field? The main one was: the field is in a very vigorous state. Fields like molecular dynamics and molecular graphics are only about forty years old, and one could imagine that such fields are on the wane at this point in their histories. "But that's not the impression that anybody apparently has, it's rather the opposite," Schulten exclaims.
While Schulten believes that the field of modeling may be very strong right now, he sees much work that needs to be done. For example, developers have to make force fields more realistic. Also, Schulten sees a need to overcome the time barrier, for now most people can only reach 10-100 microseconds of simulation time. For this, Schulten believes, developers will have to consider advanced sampling going forward. And there's more information coming from the experimental side, which needs to be rationalized. However, with all these challenges come opportunities to make key gains in understanding the life sciences. As Schulten summed up, "I think that the field has great chances to strive further, better than it ever had."


