NAMD 2.13 New Features
Improved GPU Performance
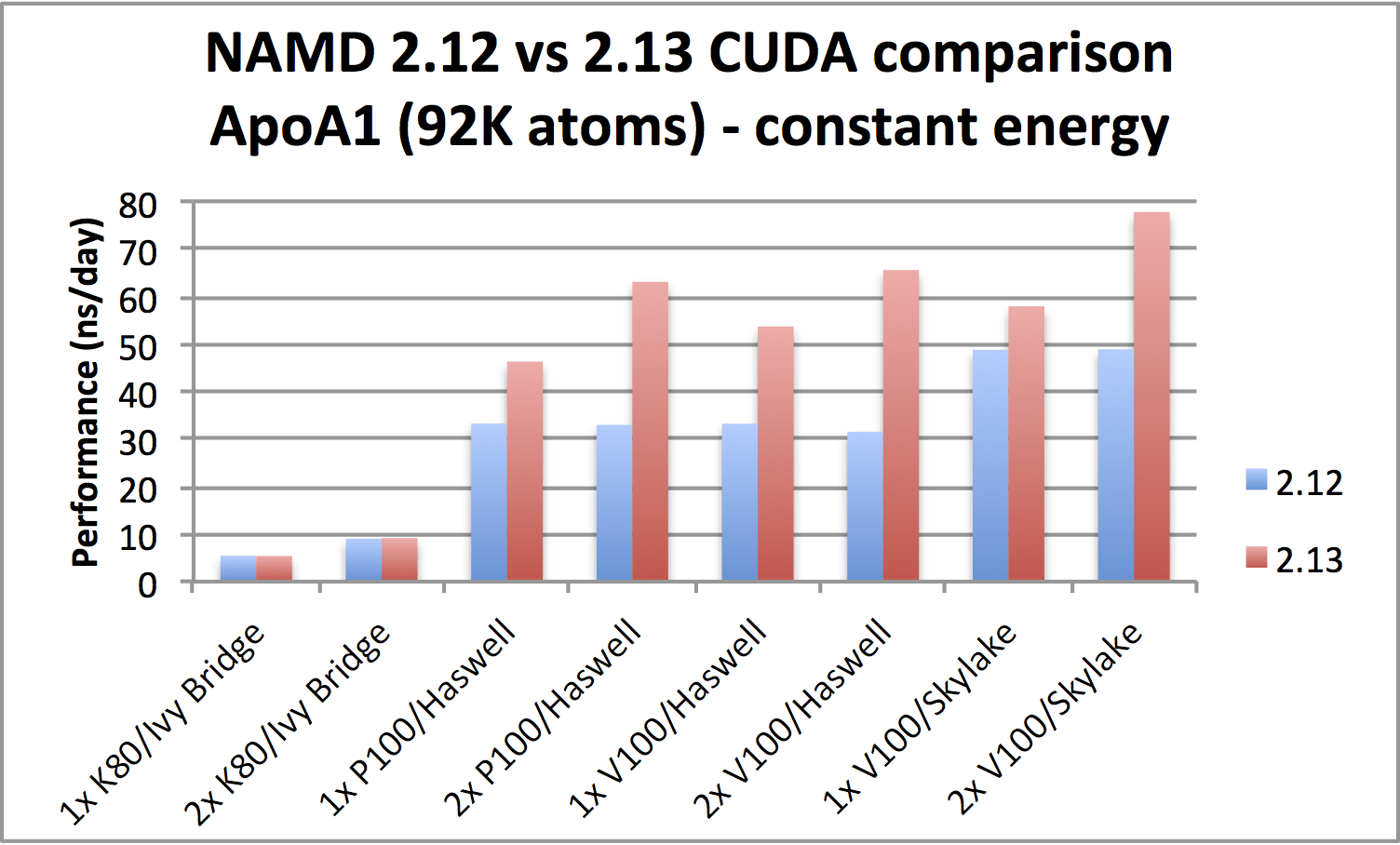
GPU-accelerated simulations up to twice as fastPerformance is markedly improved when running on Pascal (P100) or newer CUDA-capable GPUs. Benchmarks show that the new version is twice as fast for certain hardware configurations and can benefit from using a second GPU. Best results generally require also using all available CPU cores, where there is noticeable improvement in newer, faster CPUs, say, going from Haswell to Skylake. The improved performance comes from now offloading all force calculations to GPU. Note that the gridded part of PME is calculated entirely on the GPU only for single workstation simulation or when running a distributed parallel job using no more than four nodes. More discussion of performance is available on our benchmarks page. |
|
Summary of GPU support improvementsSupport for non-orthogonal periodic cells. The "useCUDA2" and "usePMECUDA" kernels introduced in NAMD 2.12 have been enhanced to support non-orthogonal periodic cells. New kernels for bonded force terms. Added CUDA kernels for bonded force terms and exclusions. CUDA support for advanced features. Extended CUDA support to now include more of NAMD's advanced simulation features, including REST2, aMD/GaMD, and Drude (without non-bonded Thole). |
CUDA 9.x compatibility. Updated all CUDA kernels for version 9.x compatibility required by Volta. Multi-GPU support. Improved CUDA device management to provide better multi-GPU support and fixed various CUDA-related bugs. NAMD now requires CUDA 8.0 or greater, Kepler or newer GPUs. Includes kernels compiled for Kepler, Maxwell, Pascal, and Volta GPUs. Support for Fermi GPUs was dropped in NAMD 2.12. |
New and Improved Methodologies
Stochastic velocity rescaling thermostatThe stochastic velocity rescaling thermostat due to Bussi, et al., is a low cost extension to the classic Berendsen thermostat that correctly samples the canonical ensemble. Unlike Langevin damping that adds a stochastic friction term to the equations of motion, this approach adds a stochastic term only to the velocity rescaling. In doing so, the method preserves holonomic constraints and zero center-of-mass momentum, making it less disruptive to dynamics than Langevin damping, while maintaining compatiblity with NAMD's constant pressure barostat. Less CPU calculation is required, which can result in up to 20% performance improvement for CUDA-accelerated runs employing modern GPUs. Contributed by Brian Radak. |
Replica exchange solute scaling (aka REST2)Replica exchange solute tempering (REST) is a powerful sampling enhancement method, that is highly transferable and provides higher efficiency than traditional temperature exchange methods. The second generation of REST, replica exchange solute scaling (aka REST2), improves sampling efficiency over the original method by scaling the intramolecular potential energy of a protein to lower barriers separating different confirmations, effectively "heating" the protein's bonds and interatomic interactions. The NAMD implementation of REST2 is made more efficient by rescaling the force field parameters for the affected atoms. In particular, this parameter scaling approach makes the method compatible with the existing GPU and vectorized CPU force kernels. The NAMD implementation is especially flexible, providing independent scaling controls over the electrostatics, van der Waals, and bonded energy terms. Contributed by Wei Jiang and Jonathan Thirman. |
Hybrid QM/MM simulationHybrid quantum mechanics / molecular mechanics (QM/MM) simulations allow the combination of precise QM calculations for regions such as active sites of enzymes, with fast classical calculation for the rest of a biomolecular system. In order to combine current QM capabilities to the scalability and advanced methods present in NAMD, a new QM/MM interface has been developed and natively prepared to make use of MOPAC and ORCA, leading QM packages specialized in semi-empirical and ab-initio/DFT techniques, respectively. A scripted interface is provided to facilitate the use of new and personalized QM methods for NAMD, allowing advanced users and developers to quickly combine and apply new methods. The QwikMD toolkit in VMD has been extended to help users prepare QM/MM simulations, providing an easy access for beginners and speeding up laborious tasks for advanced researchers. Contributed by Marcelo Melo, Rafael Bernardi, and Till Rudack. |
|
|
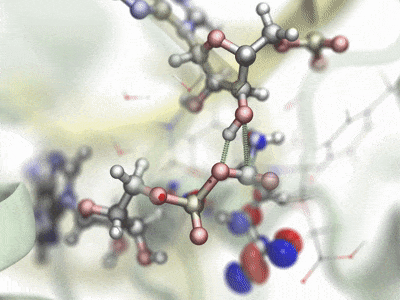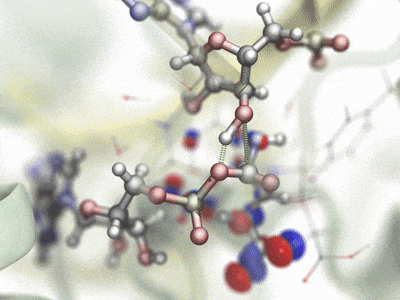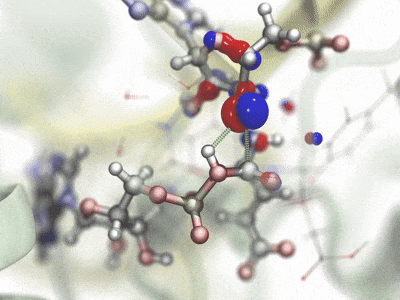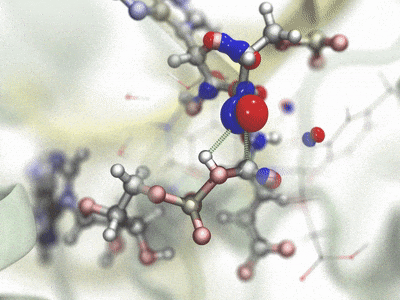
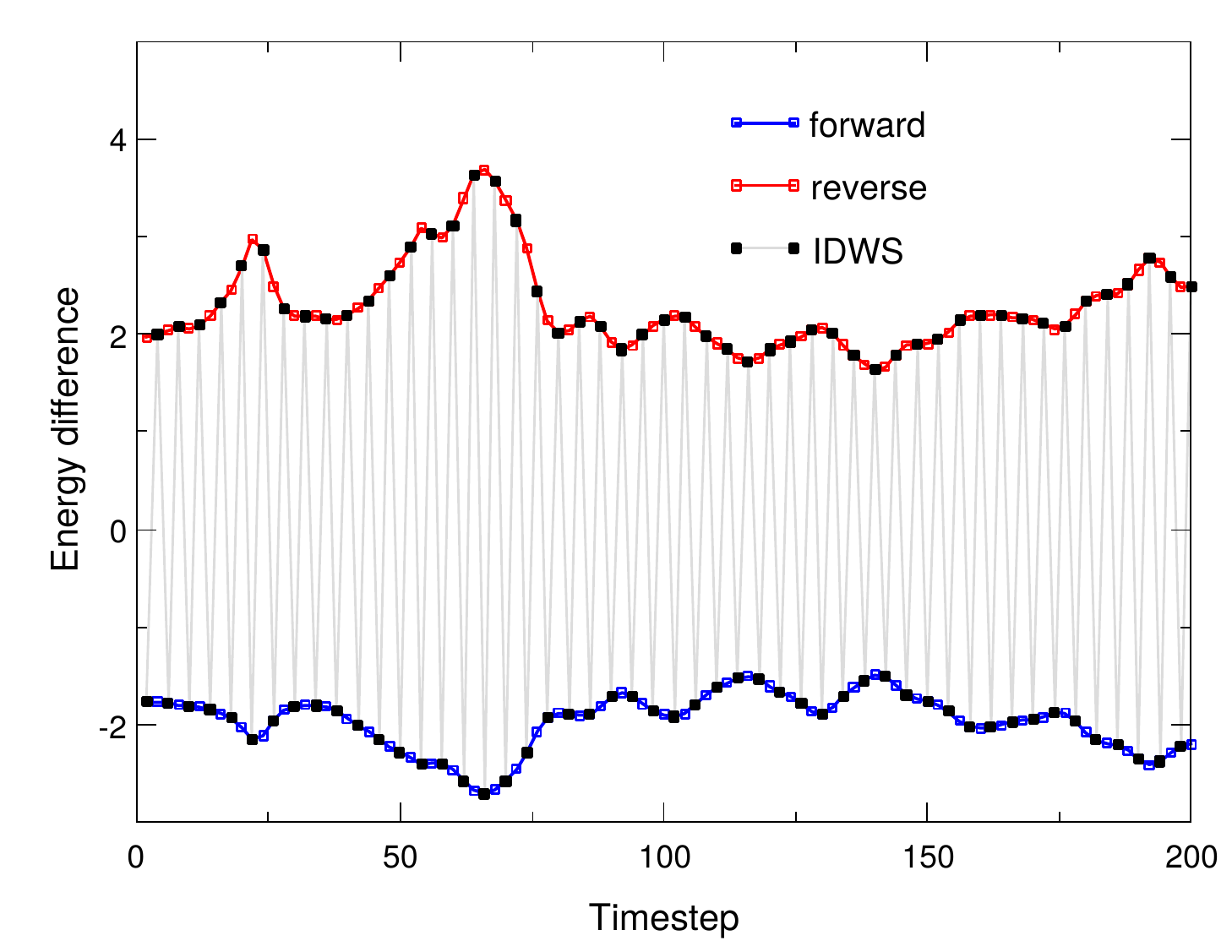
Interleaved double-wide sampling for alchemical FEPWhen doing alchemical free energy perturbation (FEP) calculations in NAMD, there is now an option to simultaneously calculate both foward and backward energy differences (double-wide sampling) at no additional cost, by alternating between forward and backward Δλ values. The alchemy output file can be post-processed to produce separate files for the forward and backward samples, which are suitable for obtaining BAR estimates of free energy using, e.g., ParseFEP. This gives a much more reliable free energy estimate than just a foward transformation and is nearly twice as fast as a sequential round-trip (forward and reverse) simulation. The updated FEP script (lib/alch/fep.tcl) makes running interleaved double-wide sampling (IDWS) just as simple as a standard FEP simulation. Contributed by Jerome Henin and Grace Brannigan. |
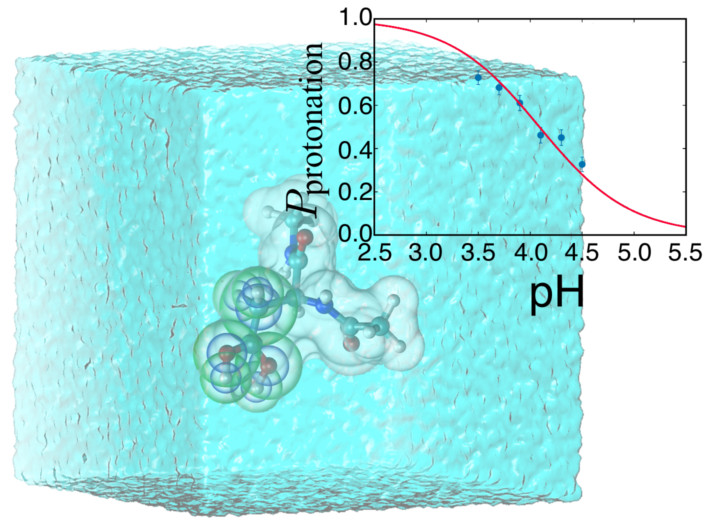
Constant-pH MDA new constant-pH MD implementation has been developed utilizing recent innovations in hybrid non-equilibrium MD/Monte Carlo (neMD/MC). The approach has three key advantages:
|
|
Gaussian accelerated MD (GaMD)GaMD aims to reduce energetic noise in the post-processing of the accelerated molecular dynamics (aMD) simulation results by making use of harmonic functions to construct a boost potential applied to smooth the potential energy surface. While effectively enhancing the sampling of a configurational space, the use of a boost potential that follows near-Gaussian distribution in GaMD is shown to improve the accuracy for the reconstruction of free energy landscapes. Example configuration scripts are available. Contributed by Andrew Pang and Yi Wang. |
τ-Random acceleration MD (τRAMD)τRAMD is an enhanced sampling protocol based on the random acceleration molecular dynamics (RAMD) method used in probing ligand egress (and access) pathways from buried binding sites in proteins. During an MD simulation, an additionally randomly oriented force is applied to the bound ligand to facilitate its dissociation. Automatically, the direction of this force is reassigned randomly if the ligand's movement in a defined time interval falls below a specified threshold distance. Whereas the original RAMD uses acceleration (kcal/mol-angstrom-amu) as the input parameter, τRAMD uses the force (kcal/mol-angstrom), providing a direct protocol for ranking drug candidates by their residence times. An R script for statistical analysis of the dissociation times of the ligands obtained from RAMD simulations, which yield their relative residence times, is available in the τRAMD package. Contributed by Stefan Richter and Rebecca Wade. |
Additional improvementsImproved lone pair support. Include support for colinear lonepairs. Improve CHARMM force field compatibility by automatically detecting lonepair section of PSF, plus various bug fixes. Contributed by Brian Radak. Extend nonbondedScaling to LJ correction term. Contributed by Brian Radak. Collective variables module improvements. Contributed by Giacomo Fiorin and Jerome Henin. |
IMDignoreForces option for QwikMD. Blocks steering forces while still allowing pause/resume/exit commands. Various Psfgen improvements. When reading topology files, recognize READ statements even if previous END statement is missing, and warn and ignore on self and duplicate bonds. For topology file PRES (patch) entries support ATOM record with name but no type or charge to specify subsequent atom insertion order. Extend segment command to query charge. |
New Platforms and Larger System Sizes
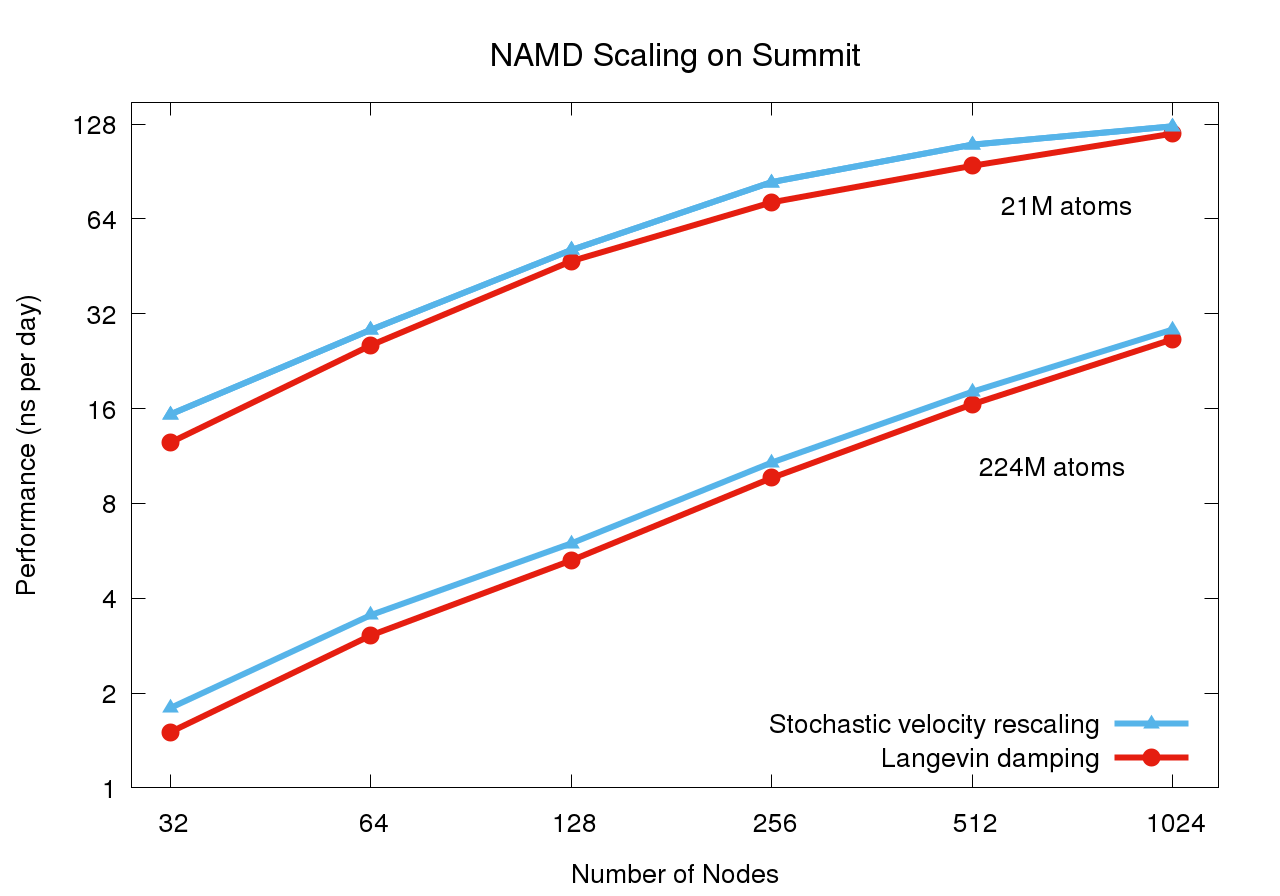
Scaling on Summit supercomputerThe Summit supercomputer newly installed at Oak Ridge National Laboratory now leads the TOP500 List ranking the fastest computers in the world. Comprised of IBM POWER9 CPUs and NVIDIA V100 GPUs, this platform poses a challenge for scalable MD simulation due to the huge increase in FLOPS provided by the GPUs while the CPU core to GPU device ratio has decreased from past supercomputers (e.g. Titan and Blue Waters), now down to just 6:1. (Although there are technically seven cores per device, one must be reserved for communication.) Porting NAMD to run effectively on Summit necessitated the offloading of the force calculation along with the updates required to support CUDA 9. NAMD's performance also benefits from the Charm++ runtime system direct use of the low level PAMI messaging layer special to IBM platforms. For constant temperature and constant pressure simulation, additional performance gains, up to 20%, are available through the use of the stochastic velocity rescaling thermostat, which reduces the CPU calculation required by Langevin damping. These and other considerations are discussed in an upcoming paper. |
|
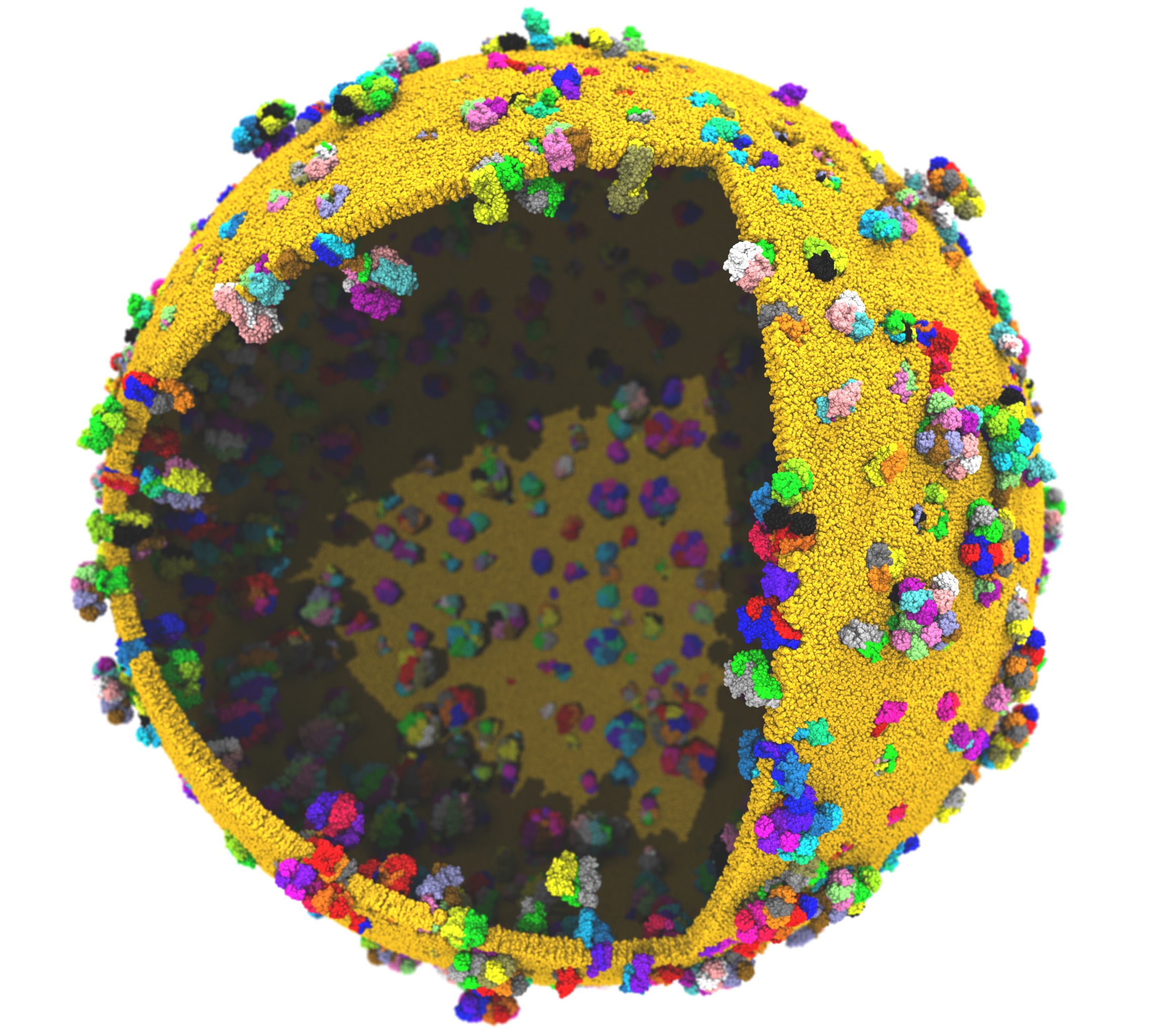
|
Support for billion-atom systemsIn order to support investigation of cell-scaled structures, NAMD capabilities have been extended to allow simulations of up to two billion atoms. Doing so has required fixing overflows in 32-bit integer indexing, issues with structure compression and the memopt build, and problems in the Psfgen structure preparation tool. The improvements have enabled initial simulations of the one billion atom protocell on Summit. Additional improvementsUpdated to Charm++ 6.8.2. Provides various bug fixes and performance enhancements. Improves support for IBM POWER 9 platforms (Summit and Sierra). |


