Historical Series: NAMD
Fashioning NAMD: A History of Risk and Reward
By Lisa Pollack
August 2012
Revised August 2013
See other related articles by L. Pollack
Download the following article, PollackNAMD.pdf
With a homemade parallel supercomputer in his backpack, Klaus Schulten waited patiently in Chicago at the O'Hare airport, hoping for no trouble getting through customs after his flight from Germany. It was the summer of 1988 and Schulten was about to start a new job at the University of Illinois. It was also the height of the Cold War, the ultimate peak in U.S-Soviet tensions, and supercomputers were causing the Reagan administration much consternation. Although Reagan had escalated the arms race and all its accompanying technological advances while in office, he wanted to keep burgeoning supercomputer developments out of the hands of the Soviets, for fear they would build better weapons.

Photo of the homemade supercomputer Klaus Schulten transported in a back pack.
The Reagan administration not only wanted to restrict Soviet scientists from running simulations on supercomputers, it also did not want Soviet scientists near a supercomputer for fear they might learn how to build their own. But in 1987 two young physics students in Munich embarked upon just such a mission to build their own parallel supercomputer, although neither had formal training in this field. Not only did they figure it out, but their project cost came in around $60,000, much less than the $17.6 million retail price tag of the Cray-2, a popular supercomputer in the late 1980s. Their advisor, Klaus Schulten, chose to risk all the grant money he had on the project, even though he had no guarantees it would succeed and even though he was not an expert in parallel computing. While the parallel computer would make possible very large simulations, Schulten sums up why he agreed to the precarious plan: “I believe in people, and when I believe in people I let them go.”
This essay looks at how a few scientists adopted a fundamentally new technology, parallel computing, to further the quest to describe large molecules. Their efforts would result in computer programs that detailed the motions of these molecules over time, and essentially fashion, as Schulten coined, the “computational microscope.” Because these scientists did not always have access to powerful computers, this history documents the measures many of them took to run the calculations they needed. But this is also a story about risk; many of the scientists put everything on the line to do this work they deemed critically important, and their decisions made their professional lives subsequently difficult but ultimately rewarding. This synopsis charts a few key players in the computational microscope history, and looks at how their risk-taking fueled the growth of scientific discovery decades later.
Abandoning a Rigid Viewpoint
In the mid-1980s Klaus Schulten was a professor at the Technical University of Munich and he had a wish list. He wanted to simulate on a computer the behavior of the photosynthetic reaction center over time. A shift in how scientists viewed these large molecules of life began late in the previous decade, along with an advance in how they could compute properties of these biomolecules, and Schulten wanted to run a massive calculation embracing this relatively new computational method. He wanted to run something called a molecular dynamics simulation, in which the internal atomic motions of these large biomolecules would yield information about the properties of the molecules. In other words, he could gain information about the function of such large molecules in biochemical reactions, by mapping out, via computer, the motions of the many atoms that make up the biological macromolecules.
College chemistry courses sometimes have, in addition to textbooks, required molecular building kits. Students in these courses make atoms, or little colored spheres, connect to other atoms via rigid, skinny sticks, almost like Tinkertoys for chemistry. The result is a molecule that can perch on the desk, be visualized in 3D, and by all appearances, is motionless. Before 1977 this static view of a biomolecule like a protein was still common in some, but not all, research circles. Something precipitated this shift in the conceptual understanding of the researchers who still believed proteins were rigid. Also, 1977 marked the introduction of a computational method that elucidated the motions of the atoms that made up biological macromolecules. This shift, toward viewing biomolecules as dynamic and toward using computational methods to mirror such dynamics, would be the first step in legitimizing the computational microscope and provide the impetus that motivated Klaus Schulten to build his own supercomputer.
Setting the Stage for Molecular Dynamics
By 1968 Martin Karplus was a very successful theoretical chemist, working at Harvard, when he realized that it was time to return to his first love, biology.
When Karplus was just a boy, he was given a microscope by his father, which helped foster a love of nature. This predilection for the natural world was only furthered when Karplus attended a lecture at the Boston Public Library entitled “Birds and Their Identification in the Field,” and he became a convert to birding. In fact, while in high school, Karplus was a co-winner of the Westinghouse Science Talent Search for his project on alcids, a family of birds. His love of biology continued into his undergraduate days at Harvard, for he wrote in his 2006 autobiography article, “I had concluded that to approach biology at a fundamental level (to understand life), a solid background in chemistry, physics, and mathematics was imperative, and so I enrolled in the Program in Chemistry and Physics.” However, his initial graduate school foray in biology did not work out*, and he moved to chemistry and received his Ph.D. in the fall of 1953 from Caltech.
(*For the full story behind the Karplus turn from biology to chemistry in graduate school, see his autobiography: Martin Karplus, “Spinach on the Ceiling: A Theoretical Chemist's Return to Biology,” in The Annual Review of Biophysics and Biomolecular Structure, vol. 35, (2006), p. 16.)
Fifteen years later, after much success in theoretical chemistry research, Karplus was ready to renew his interest in biology. He settled on the Weizmann Institute in Rehovot, Israel as the place where he could read in the famed library and discuss ideas in Shneior Lifson's group, and took a leave of absence from Harvard in order to retool his research interests and focus.
His stay at the Weizmann Institute in 1969 gave Karplus what he was looking for, and he returned to Harvard with topics to explore, namely, the origin of hemoglobin connectivity, the study of retinal in vision, and protein folding.
By the early 1970s, with many biological topics under consideration, one of his students, Bruce Gelin, developed a computer program to calculate the energy and forces of a protein simply from its amino acid sequence as well as its positional coordinates (obtained from X-ray structure experimental data). This code was used in Gelin's primary work on hemoglobin, and then also to study the bovine pancreatic trypsin inhibitor.
With the program of Bruce Gelin's now in existence, the stage had been set by the mid-1970s for the Karplus group to apply molecular dynamics to biomolecules. Andrew McCammon joined the group in the mid-1970s and set to work with Gelin's code, which already provided forces on the atoms of a protein, and used Newton's equation to calculate the dynamics, or the individual motions of the atoms of a protein.
However, there was not much affirmation from his peers for this new application of molecular dynamics to biomolecules. As Karplus wrote in his 2006 autobiography, “When I discussed my plans with chemistry colleagues, they thought such calculations were impossible, given the difficulty of treating few atom systems accurately; biology colleagues felt that even if we could do such calculations, they would be a waste of time.”
|
"Biology colleagues felt that even if we could do such calculations, they would be a waste of time" Martin Karplus |
Not only did Karplus know about the work in the 1950s on molecular dynamics, he knew that in the mid-1960s and mid-1970s other researchers had used molecular dynamics to simulate liquids. So between the trajectory calculations and the liquid calculations, Karplus decided to risk doing molecular dynamics calculations on a biomolecule even if his peers in biology and chemistry were not exactly encouraging. But prevailing negative attitudes were not all that Karplus and his collaborators were facing. These molecular dynamics calculations they wished to do needed serious computer power in order to run in a timely manner. Karplus states that in the late 1970s it was difficult to obtain computer time to do such big calculations in the United States, as most large computers were strictly for defense work. But when an opportunity to participate in a workshop in Europe promised access to a computer there, Bruce Gelin and Andrew McCammon worked tirelessly to prepare a program to run molecular dynamics on a small protein.
Their time at CECAM (Center Européen Calcul Atomique et Moléculaire) on the large computer was successful, and the three, McCammon, Gelin and Karplus, soon published their work on molecular dynamics of the bovine pancreatic trypsin inhibitor, a small protein. Since molecular dynamics literally reveals the internal motions of the protein over time, they were able to run the calculation for 9.2 picoseconds of simulation time. Basically the molecular dynamics program had to run for at least the time it takes for a biological process to unfold. Although a few picoseconds may seem like a small amount of time to elucidate behavior of a protein, this would be one guiding force in propelling future investigators to seek out larger and faster computers. Scientists wanted to extend the simulation time of a molecular dynamics calculation, i.e. utilize the computational microscope for durations beyond the picosecond range.
Computing Schulten
Schulten was aware of this work of McCammon, Gelin, and Karplus because he was finishing his graduate studies at Harvard, where Karplus was one of his advisors, and McCammon and Gelin were fellow students who sat right down the hall at the beginning of their molecular dynamics project. The 1977 paper had a definite impact on Schulten, who was trained to use theoretical and mathematical methods such as those employed in chemistry and physics. “I realized that this computational approach,” he says of molecular dynamics, “opens new doors to describe problems that you couldn't do with a purely theoretical approach that I had taken until that time.”
But embracing a computational method instead of staying exclusively with pure theory had its costs, and he soon became known as “Computing Schulten.” “I paid a big price for it, because basically during much of my career people thought I was stupid,” reminisces Schulten. “Although I continued publishing mathematically-oriented papers, for every computing-oriented paper I had to redeem myself with ten theoretical papers, and I couldn't possibly do that.” Schulten would not be the only one in this story who would be denigrated by his peers for moving away from pure theoretical research.
In fact, selling the usefulness of the computational microscope, or the molecular dynamics approach, in its very early stages was also a battle Schulten sometimes had to wage. In 1985, while still a professor in Munich, Schulten went to a supercomputing center in Illinois to run some calculations, and returned to Germany with a movie illustrating a protein in motion, based on molecular dynamics simulations. When Schulten showed the movie, one of his colleagues became quite enraged. “He got so upset when he saw it, he almost wanted to physically attack me,” Schulten recounts. “He told everybody this is the greatest rubbish he'd ever seen in his life. He was a crystallographer who thought basically of proteins as some kind of Gothic cathedral that were cast in stone.”
Despite the many struggles Schulten had to face, he was intent on using molecular dynamics for his work; he was sure it would lead him to new discoveries that would be valuable for science. “My love of scientific discovery,” Schulten confirms, “made me do the dirty business of computing.”
A Perilous Plan
This was exactly what motivated Schulten in 1987 to make an audacious judgment call. He really wanted to thoroughly understand photosynthesis, in which sunlight is transformed into biochemically exploitable energy. But an important macromolecule in the process is a large protein that sits within a membrane. Simulating the protein by itself and neglecting the membrane and liquid that surrounds it was not highly desirable because such isolation is not a natural environment for the protein. This protein is about 12,000 atoms and the membrane and water that surrounds it in its natural environment adds another 100,000 atoms to the tally. In the late 1980s no supercomputer was even close to capable of handling that task. Schulten thus decided to focus on understanding and simulating just part of a membrane in water and managed to run a calculation on a Cray-XMP but it only covered a few picoseconds of time and it taught him one thing: he needed a supercomputer all to himself, perhaps for a year or more, to really understand the mechanism.
While Schulten, teaching physics at the Technical University of Munich, was wondering how he could commandeer a supercomputer all for himself, a young student at the same university was wondering how he could build faster computers. Actually, Helmut Grubmüller knew in theory how to make a computer faster, but was really wondering if he could get someone else to pay for it. Enrolling at the university in Munich in 1985, barely twenty years old, Grubmüller recounts how his choice of specialty shaped his goals: “When I started physics, I quickly saw that in order to learn more about how nature works, a computer can help tremendously. That's ultimately the reason why I was so fascinated with computers; you could do calculations which you couldn't do otherwise.”
His fascination prompted him to begin soldering together a multi-processor computer in his second or third semester, one that would be much faster than what was available to regular students in those days. Although it was technically a parallel computer, Grubmüller just called it a multi-processor at that point. It was the mid-1980s and there was no World Wide Web to consult for details about how to construct his own. Instead he read any books about microprocessors he could find, and talked to company representatives on the phone about technical details of the parts they sold. He cites as most important for making headway the data sheets of the chips he used as the processor, which were Motorola's 68,000 line. But he could only get so far on his own. “That very quickly blew my budget,” recounts Grubmüller, “My private budget was a thousand dollars or so.”
His persistent desire to build a faster computer led him to concoct an admittedly naïve idea. “I approached some of my professors and simply asked them if they would be willing to conduct a deal,” Grubmüller says. “I would build a larger machine, larger computer, and they would pay for the hardware.” Most of his professors told him that was crazy and rejected the idea right away.
Klaus Schulten was teaching Grubmüller a physics course in mechanics at the time, and he remembers the day Grubmüller approached him with his homemade multi-processor computer. Grubmüller asked Schulten if he could visit his office, and Schulten had just been expecting they would have a discussion. “He opened his bag and got this out and showed it to me,” recalls Schulten. “He showed me the results and I fell almost off my chair.” With the demonstration of some of the capabilities of his multi-processor, Schulten was impressed. “So I immediately integrated him in my group,” says Schulten. Schulten told him there were other people in the group that also knew lots about computers.
One of those people was Helmut Heller who, Schulten would learn in dramatic fashion, was infamous at the university. When just in high school, Heller started teaching himself about computers. He had one of the first personal computers, the PET 2001, introduced by Commodore in the late 1970s. It had a mere 8 kilobytes of RAM and a cassette tape for archiving data. “I started off with learning BASIC and writing BASIC programs on that machine,” recollects Heller. “Then soon after I went into writing machine assembly code.” He had some friends in his hometown who were equally intrigued with computers and they taught each other as well.
When he entered the Technical University of Munich, he continued using computers, sometimes even employing them to go beyond what was required by his professors. This is how he grabbed Schulten's attention. Heller was enrolled in a physics class taught by Schulten. According to Schulten, the second half of that semester consisted of seminars by the students on small computer projects. One day Schulten was going to teach as usual when he thought he had entered the wrong classroom.

Klaus Schulten, Helmut Grubmüller, and Helmut Heller in the late 1980s. In the middle is one of ten boards comprising their homemade parallel computer T60.
Apparently Heller made a complicated fractal movie on his computer for the assignment. “He showed us how he modified the computer to do this calculation,” Schulten relays. “Basically he went into this computer with wires and so, even today I couldn't tell you what he really did.” Watching Heller toy so smoothly with the computer really left an impression on his professor. Schulten was pleased to invite Heller to join his group.
“Sometimes you take your chances,” says Schulten on his decision to let Grubmüller and Heller build a parallel supercomputer. “And so that's what I did. And to this day I don't know if I would do it again.” The risks were enormous for Schulten. Although he would have a supercomputer all to himself for a molecular dynamics simulation, if it somehow didn't work out, he would have nothing to show. He was not an expert in parallel computing at that time and had little basis for assessing the project's feasibility. Moreover, he would have to explain to the funding agency how he had sunk tens of thousands of Deutsche Marks into a failed computer when the money was earmarked for equipment that usually came with warranties and service contracts.
Creativity on Display
Heller and Grubmüller, however, made a great team, even though what they were trying to do was no small feat. Not only did they have to build a supercomputer, but they also had to write software to work in parallel on it. “Helmut Grubmüller and I, we had very similar but also complimentary computer knowledge” recalls Heller. “He was more on the hardware side and I was more on the software side.” However, they designed both the hardware and the software together, consulting each other at every step. “It might have been that's the only way this worked so well,” says Schulten. “Maybe one personality alone would have not been good enough to do it.”
Schulten was amazed by their ingenuity. “They thought in terms of solutions rather than difficulties,” he recalls. For example, when the pair told Schulten they needed an oscilloscope to test that the computer was working properly, Schulten knew he didn't have the additional money for it. But the two students assured him that if they could find two or three oscilloscopes on the equipment trash heap, they could assemble one working oscilloscope out of parts, and that is what they did.
The pair decided to build their parallel computer, eventually named T60, out of processors known as Transputers, because purportedly they were as easy to assemble as Lego blocks. The final computer had 60 nodes. There were ten circuit boards, with six Transputers per circuit board. Grubmüller and Heller even designed the PCB board layouts.

One of ten circuit boards that made up T60. The six Transputers on the board are clearly visible.
Since soldering each of ten circuit boards was time consuming, the other members of Schulten's research group pitched in. Each member got the parts and an instruction sheet and soldered a board, which was at least a six-week job, and then signed their names on it. “It really showed the research personality of a student,” claims Schulten. “This kind of job that everybody in the group had to learn really reflected very closely how they actually performed later in science.” In fact, Schulten feels that this soldering project was even better than traditional exam results in delineating which candidates would make good graduate students to accept in the group.
Heller recalls the tribulations the team faced in writing their code for a parallel machine, a code they named EGO. “I remember that when I first had EGO do parallel computations that scaled well, I was very excited, but soon I found out that the results were all wrong! I had a fast parallel program, giving wrong results.” Finding the reason for this was not easy. Schulten bought a hunk of industrial marzipan, which he knew was Heller's favorite, and cut off slices for him every so often to encourage him to keep at it. After much good-natured teasing by others in the group for his fast but wrong calculations, Heller eventually figured out he had to properly synchronize the parallel tasks, as he had given too much freedom to the tasks at first; the solution took many days and long evenings of work.
Transporting a Supercomputer during the Cold War
The project, to build and program the T60, began late in 1987. In the middle of its completion, Schulten took a new job in the United States, at the University of Illinois at Urbana-Champaign. Instead of shipping the T60 over to the United States, and waiting weeks for it to arrive and then go through customs, Schulten decided to just carry the computer in a backpack due to its portable size.
During the Cold War, the United States had long been at odds with its western European allies over the issue of restricting high technology items, like certain computers, to Soviet bloc countries. European nations did not want their commerce and profits with the Soviet bloc curbed by the United States, and even computer companies in the United States were worried that their overseas business would be curtailed by government restrictions.

The elegant wiring of T60, a supercomputer Klaus Schulten spirited through customs in Chicago in 1988.
Not only did the Reagan administration want to restrict businesses, both domestic and foreign, from exporting supercomputing technology to the Soviets, it also wanted to keep Soviet scientists out of its newly formed supercomputing centers. In 1985 the National Science Foundation awarded $200 million in grant money to four universities to open supercomputer centers on their campuses. The move was deemed necessary, and authorized by Congress, to give academic researchers access to supercomputers that were usually restricted to defense work done by the Pentagon and National Laboratories. As one of the key scientists who lobbied the U.S. government said, of using standard computers instead of restricted supercomputers, ''[it's] like riding in a horse and buggy while jets are flying overhead.'' But in 1986 the Reagan administration proposed prohibiting access to these supercomputer centers by Soviet scientists. Such was the climate in summer of 1988 when Klaus Schulten decided to take his homemade supercomputer onto a transatlantic flight and carry it through customs in Chicago.
Schulten was aware that, because of its data processing rate, the T60 computer had to be registered, especially when being exported or imported. He approached the custom's officer and showed him the supercomputer.
“What is this?” asked the customs officer.
“This is a computer,” replied Schulten.
“Why are you showing it to me?”
“This is a very powerful computer and it needs to be registered with you.”
Schulten then explained some technical details of the machine to the officer. Finally the custom's officer looked at Schulten and said, “Please, just put it back and go.”
Tribulations of Theoretical Biology
When Schulten arrived in Illinois, his students finished building the T60 and programming it. The program, named EGO, was written in OCCAM II, a language around which the Transputer had been designed. Just as building the T60 required major efforts, writing code that ran in parallel was an equally arduous task. One of the key indicators in parallel programming is how well the software scales; scaling well means that the run time for a calculation speeds up when more processors are used. For example, a human endeavor that scales well would be clearing a tree of its apples. As more people are shuttled in to help pick the apples, the time it takes to clean the tree decreases.
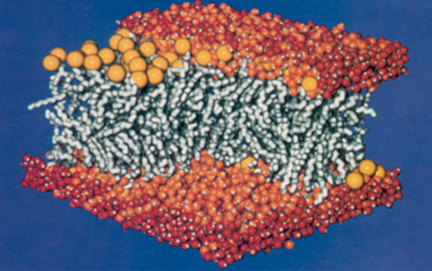
Membrane structure, simulated on T60 for twenty months, encompassing 27,000 atoms. Image from Heller et al., 1993.
With such incredible efforts applied to the membrane problem, you can imagine Schulten's surprise when the article on it, with co-authors Helmut Heller and Michael Schaefer, was rejected within a week, even though the results agreed well with experiment. The paper contained theoretical biophysics results. Schulten acknowledges that to publish theory related to biology is very tricky, especially in high-impact journals. He says even today he has a better chance of acceptance if he teams up with experimentalists. In his 2006 autobiography article Martin Karplus summarizes the impediments theorists face when publishing biology-related results: “The problem is almost as prevalent today as it was then, i.e., if theory agrees with experiment it is not interesting because the result is already known, whereas if one is making a prediction, then it is not publishable because there is no evidence that the prediction is correct.”
The mystery of the rejection was never cleared up. “I still to this day don't know why it was rejected,” says Schulten. “I was so furious that even though I published many papers in this journal, I told them I would never publish there anymore.” Heller and Schaefer and Schulten eventually published in the Journal of Physical Chemistry, a paper that is now highly cited.
This foray into molecular dynamics on such a then-mammoth system provided an early justification for the use of the parallel computer as computational microscope. Membranes are important since they act as cell walls, and cells in higher species are divided into compartments by membranes as well. It was important to take a large membrane segment for analysis because real cell membranes are continuous and don't have an edge per se; a small piece of membrane is an artificial system. Other types of conventional microscopes could not capture some of the critical features of a membrane that molecular dynamics elucidated. “When you look at it in a moment,” says Schulten of a membrane, “you see it all fluid and moving. And that's a little bit difficult to get with all these other microscopies. It's not a little bit, it's impossible to get.” When the membrane calculation was completed, Schulten evaluated if his risky foray with the T60 produced a successful computer simulation, and the answer was a resounding “yes.” “We could actually compare with lots of experiments that were not seeing the detail that the computer saw,” Schulten recollects. “They measure average properties that gave us a very good indication that what we actually saw with the computer was a correct picture.” The successful computation only whetted Schulten's appetite for studying larger and larger systems. Little did Schulten realize that his thirst for massive systems on parallel machines would lead to a student rebellion of sorts, and a software product called NAMD that would define his career.
Seeking Collaborators
In 1989 Schulten founded the Theoretical Biophysics Group (which would later become the Theoretical and Computational Biophysics Group) at the Beckman Institute of the University of Illinois. He received a two-year grant from the National Institutes of Health in 1990 for this, more or less an initial grant to get his center up and running. By this time, Schulten knew a thing or two about parallel computing, compared to being a relative neophyte on the matter when he had sunk all his research money into a parallel machine back in Munich; he realized he could study massive biomolecules by taking advantage of parallel machines. There were already software codes, some even freely available since the mid-1980s, that did molecular dynamics, but Schulten realized that his needs far exceeded their capabilities.
In fact, Schulten realized his needs exceeded even his own capabilities as a computational biophysicist if he wished to carry forward his plan to study more massive biomolecules in their natural environments. Schulten decided to step outside his field. “In my opinion that was some foresight on his part to actually look for computer scientist expertise,” recalls Laxmikant “Sanjay” Kalé, one of the scientists Schulten approached. Motivated by three reasons, Schulten's judgment compelled him to cross disciplines and seek help from the computer scientists at his university.
First, up to 1991, the programming for molecular dynamics in Schulten's group had always been done by his physics students. After the two Helmuts wrote EGO for the home-built computer, another student, Andreas Windemuth, wrote a molecular dynamics code for a parallel computer, the Connection Machine.
|
"In my opinion that was some foresight on his part to actually look for computer scientist expertise" Sanjay Kalé |
Second, Schulten cites that he realized programming techniques were changing, and especially that FORTRAN, developed in the 1950s, might not be his top choice as other languages started to gain prominence. “We needed better programming languages that were more systematic,” declares Schulten.
Third, mastering code for parallel machines seemed daunting. The code for the Connection Machine didn't seem to be easily transferable to other parallel machines, and was difficult to write in general. The nuances of building a code that was portable to other parallel machines, at a time when so many different types of vendors were selling parallel computers, helped to convince Schulten to seek outside input. He wanted expertise on how to handle parallel machines, and he knew computer scientists could provide knowledge about a technology that was rapidly evolving and changing.
With the above threefold motivations in mind, and needing to write a strong renewal grant for his center, a big grant that would provide five years of funding instead of just two years, Schulten approached two computer scientists at the university in Illinois and asked them to be on the renewal proposal. One, Bob Skeel, was an expert in numerical algorithms, and the other, Sanjay Kalé, was an expert in parallel programming. In 1992 Schulten received a five-year grant from the National Institutes of Health and so began the genesis of the software code called NAMD.
Grad Students Rebel
Actually, Schulten cites the early work on T60 as a seed of NAMD–that, coupled with a student revolt that happened in the early 1990s. At this time Schulten hoped to continue using the molecular dynamics codes that had been developed for the Connection Machine and T60. Bob Skeel's graduate student, Mark Nelson, was studying the code for the Connection Machine, and Schulten's graduate students, Bill Humphrey and Andrew Dalke, were examining the code for T60. All three were vexed by the existing codes they were inspecting. “I banged my head against that code for a couple of months,” recounts Nelson, “and was going nowhere. There were no comments, and all the variable names were abbreviations of German names, which really didn't help me.”
The three graduate students, Nelson, Humphrey, and Dalke, together decided their task would be easier if they just wrote a new code from scratch. They surreptitiously worked for about a month until they had a viable code. Taking a big risk with their advisors, they presented at a group meeting their idea to create a whole new code and nix the existing ones, which were too complicated to follow. “They wanted to write it in C++,” recalls Schulten. “They thought the professor will shoot them, for being so bold and telling him NO. They even put conditions on how they wanted to go forward.” In fact, however, Schulten was thrilled at the initiative. “So I was actually very excited myself,” recalls Schulten, “because I saw here a situation that was almost like the Helmuts before them. The students were going for it. They really wanted to realize it themselves.”
At this point Schulten's group was well equipped to craft a software code from scratch, now that he had computer scientists on the team. One of those computer scientists, Sanjay Kalé, had an almost miraculous coincidence that led him to NAMD, and soon learned that he needed a thick skin for what was about to befall him.
Rejecting Platonic Computer Science
In the early 1980s, while a graduate student at SUNY Stony Brook in New York, Kalé began working on parallel logic programming. Specifically he was using the language Prolog in parallel to study artificial intelligence. “It so happens,” recounts Kalé, “if you look around in the field now, a huge number of people in parallel computing actually were doing parallel Prolog in the mid-1980s.”
When he got a job at the University of Illinois, he continued working on parallel logic programming and secured tenure in 1991. While he was doing this more or less “pure” computer science-based research, he began to ponder application of his work. “I was doing state-based search, artificial intelligence in general, in parallel, like the traveling salesman problem, etc.,” says Kalé of this time period. “But my interests had started shifting to: how can we apply this to problems that engineers and scientists have?”
In an amazing coincidence, right after Kalé received tenure and was about to go on sabbatical to India for a semester, with application now in the back of his mind, Klaus Schulten approached him to inquire if Kalé would be on the 1991 grant proposal for the center, as the parallel computing expert. Kalé suddenly had an application-oriented project right in front of him, literally in his own back yard. He spent some of his sabbatical reading about biology and biological simulation, and when he returned from India he started studying the two programs Schulten had used for parallel molecular dynamics calculations.
Kalé thought long and hard about how and why he wanted to approach an application-oriented project. “In computer science this kind of activity is just not done,” he muses about this bold leap toward application he knew he could make once he got tenure; but he was determined to take such a risk because he felt strongly about application-based work.

Sanjay Kalé, a computer scientist at the University of Illinois since 1985.
He realized firstly that computer scientists often research in the abstract. “That doesn't work, because you're not rooted in real problems,” Kalé says. “So I call it the platonic computer sciences. The idea looks beautiful, so you develop the idea, and then you throw it over the fence to the real world. And the real world doesn't care because it doesn't solve its problems.”
“If you just work for one application,” he continues, “that doesn't work because you don't develop technology that's broad enough to enable a broad set of applications. So you need to be looking at multiple applications.”
By the early 1990s Kalé had secured application-oriented projects, with Klaus Schulten on molecular dynamics and another one on fluid dynamics with a collaborator in mechanical engineering. As far as the molecular dynamics was concerned, Kalé had been studying the previous codes developed by the Schulten group. Around 1994 the graduate students confronted Schulten and asked to start over with a new code. “We decided to explicitly formally design it as a program,” recalls Kalé. “Step back and design it as a program rather than let it be organically grown.” This formal design would lead to the first version of NAMD, a prototype. The name initially referred to “Not Another Molecular Dynamics” program, which graduate student Bill Humprey coined, whimsically inspired by a compiler name of similar nature; soon the acronym came to stand for NAnoscale Molecular Dynamics. NAMD's explicit insistence on being parallel from the outset would have lasting implications in terms of the success of the software.
Although Kalé fulfilled his desire to work on applications, the last decade of the century was difficult because of it. Kalé and his graduate students had developed a parallel programming language they named Charm++, which actually became very critical and valuable for the success of NAMD. His own computer science research during the decade focused on it. “What we were proposing was a parallel C++ or parallel object-based language, which was novel in its own way and substantially a departure from how people were doing parallel programming,” recounts Kalé. “So that was my challenge, to get people to accept that.”
But he had very little success. And he has stacks of rejected grant proposals to remind him of it. The reviewers said what he was proposing was either impossible or passé. His publications were not always appreciated either: “We would write papers and I would say 'This is demonstrated in molecular dynamics.' And people would say, 'This is just parallel C++. Lot's of people are doing parallel C++.' [They were] not quite paying attention to what was a novel element. And the application was ignored anyways.”
Kalé also feels that working outside of his field was another reason he struggled. “So people pay lip service to interdisciplinary research, but in fact it's very hard to do,” he says. “And people don't respect interdisciplinary research as much in either discipline.” Looking back on that time, Kalé, like Schulten, assess the trajectory his professional life took after his decision in the 1990s to move toward application: “I still don't advise young people to do what I did unless they really understand what the risk is in that career path.” It would be years until that risk paid off for Kalé.
Early Ingredients of NAMD
Many factors shaped the purposeful design of NAMD in 1994, not the least of which was that it was now well funded by the NIH grant. The graduate students insisted on using the language C++, a relative newcomer in the field of programming. “C++ offered a nice combination of having it be object-oriented,” declares former student Mark Nelson, “but we could still write the key computational pieces in C and get the speed that we needed.” Use of C++ in NAMD made it unique among molecular dynamics codes at the time.
Since NAMD was to be tailored for parallel machines, the developers had to take into account how the many processors that made up the parallel machine would communicate with each other while running molecular dynamics calculations. Kalé and Attila Gursoy, his graduate student and later his post-doc, were working on the Charm++ environment and something called message-driven execution. It turned out that the structure of molecular dynamics as a parallel application was very well suited for what they were doing with message-driven execution. Although the technical details of message-driven execution are beyond the scope of this article, suffice it to say that Charm++ used it to effectively enable processors to talk to each other during molecular dynamics runs.
While C++ and Charm++ were unique elements to NAMD, the from scratch parallel design was a hallmark. In the early 1990s other very successful molecular dynamics codes like Amber and CHARMM (which is a Harvard product and not to be confused with Charm++, which was developed in Illinois) took a different tactic when it became apparent software needed to be developed for parallel computers.
|
"That design has stood the test of time from then till now" Sanjay Kalé |
Sanjay Kalé cites the advantages the from scratch design afforded NAMD: The software scaled well and was able to handle whatever new machines were rapidly developing on the market. “That design has stood the test of time from then till now,” Kalé reflects. “Pretty much the architecture of the parallel program has remained the same, even though at that time we were running on this HP cluster, roughly eight processors. And now we run on 200,000 plus processors.”
And the vendors of the parallel computers were taking notice. From its inception NAMD was offered to the community for free, and started accruing more and more users over the years. “When it became popular then IBM and so on started to pay attention to it a lot more,” Kalé remarks. “And the other computer scientists kind of had to pay attention to us and Charm++ because the vendors were paying attention to us.”
Rewarded After a Decade
Almost ten years after Kalé began his odyssey into the world of parallel computing application, on a path that was arduous at best, he was finally rewarded. Each year an international conference is held which is very important for computer scientists, called “Supercomputing” or more recently, simply SC. The Gordon Bell prize is given each year at the conference to recognize accomplishments in parallel computing. For NAMD and its developers, affirmation came at SC2002 with the award of a Gordon Bell prize.
While Kalé may have worked for years on applications like NAMD and the fluid dynamics mentioned earlier, he also was doing more or less pure computer science research of his own that originated from his collaborations on application. He believes that the Gordon Bell prize gave his group and Charm++ reflected glory. “In fact, I'm very grateful for that,” Kalé notes, “because our computer science ideas would have remained unappreciated, or less appreciated. And it would have been difficult to get them appreciated but for NAMD.” Because of his reputation with NAMD, Kalé now has projects in weather modeling, computational astronomy, and computational chemistry.
The Computational Microscope Comes of Age
By now the computational microscope had become sophisticated. In 1977 the first molecular dynamics simulation was done on a small protein in vacuum and lasted for 9.2 picoseconds. By 1996 NAMD was simulating a system with 36,000 atoms, the estrogen receptor with a segment of DNA in salt water, for 50 picoseconds. The calculation ran for two or three days on an 8-processor cluster. In 2004, on an aquaporin-1 system of 81,065 atoms, the calculation ran for 22.4 hours on a 128 processor parallel machine. By this time the molecular dynamics calculation on aquaporin-1 simulated 5000 picoseconds, or 5 nanoseconds of time.
With these kinds of advances the computational microscope took a foothold. “In the old days the computer simulation wasn't so accurate,” recollects Schulten. “So it would have been hubris to just say, 'This is a computational microscope.' People would have laughed at us and said, 'This is a really bad microscope you have here.'”
When asked what forces drove the computational microscope to improve, Schulten is clear that there is a deeper cause than the obvious behind his motive for continually refining NAMD to do larger systems. “We didn't become larger because I wanted to beat my chest and say I can do more atoms than you, but rather for a clear intellectual reason. And the reason is, I think it is essential for the very nature of living systems, of living cells in particular, that you need to describe how biological macromolecules, in particular proteins, assemble and cooperate.” Schulten ultimately wanted to look at not just a single protein, but at scores of proteins and the way they organize themselves and form societies in a living cell. “The key point,” Schulten continues, “is simply that these macromolecules have to find each other in the right proportion, in the right geometry, at the right place in the cell, and they have to cooperate.” And in order to describe this, NAMD had to improve to the point that it could describe tens or hundreds or thousands of proteins.
Oftentimes the computational microscope predicted correctly what experiment observed; sometimes it was ahead of experiment. One such time was an instance of using NAMD to run a “steered molecular dynamics” simulation. In steered molecular dynamics one can apply external forces to a protein and map its subsequent behavior over time. An ankyrin repeat is a certain sequence of amino acids and this sequence is found in more than 400 human proteins. In 2005 Marcos Sotomayor, David P. Corey, and Klaus Schulten studied the elastic properties of these ankyrin repeats using steered molecular dynamics, ultimately to better understand hearing and balance; a year later, in 2006, experiment confirmed their simulation results. In a highly cited Science article in 2007, “Single-Molecule Experiments in Vitro and in Silico,” Sotomayor and Schulten argue that in silico experiments, like the one studying ankyrin repeats, “have become a powerful tool complementing and guiding in vitro single-molecule experiments.” The term “in silico” refers to a study done by computer analysis. The justification of the computational microscope as a powerful tool only grew stronger as this in silico experiment preceded the in vitro experiment.
NAMD in the Twenty-First Century
NAMD got its start in the early 1990s, and the seed of it goes back to the homemade parallel computer built in the late 1980s and the program EGO that was written for that specific supercomputer. Schulten wanted to run bigger and bigger systems and so NAMD was born and then grew because it was well funded. A key player behind NAMD's success is lead developer Jim Phillips, a former graduate student of Schulten's in the 1990s. Now Schulten calls Phillips “the father of NAMD,” as Phillips has been crafting creative solutions for the parallel software for almost two decades.
Many systems have been elucidated with the help of NAMD in its roughly twenty-year history. A complete list is beyond the scope of this article; however, in the appendix appear several papers, which describe molecular dynamics on key proteins studied by Schulten's group in Beckman. Each of these publications has been highly cited. Some of the topics covered are studies of the titin protein that is responsible for passive elasticity of muscle, and examination of how Aquaporins work to pass water or glycerol through a membrane but forbid protons from crossing the same membrane. The fact that these papers in the appendix have more than a hundred citations each argues for their importance. But to round out the history of NAMD, a focus on what the computational microscope has achieved in its study of viruses illuminates the legacy of NAMD in the last few years.
It goes without saying that research on viruses will aid in combatting diseases. In 2009 the World Health Organization declared a pandemic of swine flu. Named H1N1pdm, it was quickly discovered that this strain of influenza had acquired resistance to Tamiflu, a drug that treats people recently infected with influenza.
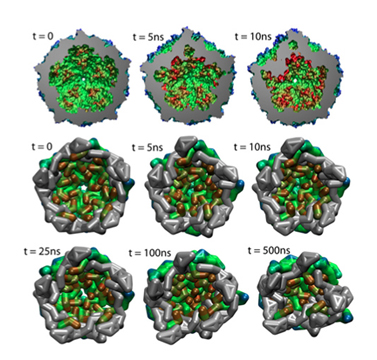
Collapse of an empty capsid. The three snapshots on top are from all-atom simulations , and the six on the bottom are from coarse-grained simulations.
The satellite tobacco mosaic virus is one of the smallest known viruses; that fact, coupled with advances in computer power, enabled simulation of this entire life form using NAMD. Researchers at Illinois teamed up with those at the University of California, Irvine, to study the stability of the full virus particle as well as its component parts in a molecular dynamics simulation. With results published in 2006, this was most likely the first all-atom simulation of an entire life form, encompassing over a million atoms. The findings established what factors are critical to the structural integrity of the complete virus and also what factors might guide assembly of the particle.
Viruses are very primitive particles and many only consist of a protein shell, or capsid, surrounding a strand of DNA or RNA. To reproduce, a virus enters a host cell and hijacks that cell's machinery to create more virus particles. While the capsid plays the role of protecting the virus, it also must somehow become unstable and release its internal components into the host cell for reproduction. Discerning the motion of the proteins that make up the capsid during such a release is highly desired and a study in 2006 examined the dynamics of various capsids. A relatively new method, coarse-grained molecular dynamics, permitted simulations of entire capsids, which until that time had been out of reach. Scientists at the University of Illinois refined the coarse-grained molecular dynamics technique and used NAMD to illustrate capsid stability for several viruses. With the new technique, it became possible to simulate capsids bigger than 10 nanometers, and look at transitions between stable and unstable structures. This work can provide useful information for battling viral diseases.
The future looks bright for NAMD. Klaus Schulten's goal of understanding biological organization, the very essence of what makes a cell a living cell, is looming closely on the horizon, after forty years of dedication and creativity. “So now I can slowly do it,” remarks Schulten on biological organization, “close to retirement. And other people behind me can use it now and can do it much better. And that makes me of course also happy.” And the computational microscope continues to elucidate larger and larger systems. “Today we have a simulation running for real science work on 20 million atoms,” Schulten says. “ And we have a simulation just to be ready for the next generation of computers, with 100 million atoms.” The combination of vision and risk taking has certainly paid off in terms of scientific discovery. From elucidating the estrogen receptor to better understand breast cancer, to illuminating viruses to better combat disease, the computational projects made possible with parallel computers will continue to push boundaries. And as long as bold scientists take great personal risks to attempt what has never been done before, and continue to re-imagine new pathways for the computer to act as a scientific instrument to assist researchers, the future of the computational microscope seems certain to bring more enlightening discoveries.
Other Related Articles
If you enjoyed this article, you might be interested in these other histories:History of Magnetoreception
History of Polyenes
History of Membrane Proteins
History of Bionanotechnology
History of Photosynthesis
History of VMD
History of MDFF (Molecular Dynamics Flexible Fitting)
Comments?
For comments or questions, Lisa Pollack can be reached at: lpollack AT ks.uiuc.eduCitation
To cite this article, please use the following:Pollack, Lisa. "Fashioning NAMD: A History of Risk and Reward." www.ks.uiuc.edu. 1 August 2012, from http://www.ks.uiuc.edu/History/NAMD
References
Martin Karplus. Spinach on the Ceiling: A Theoretical Chemist's Return to Biology. The Annual Review of Biophysics and Biomolecular Structure, 35:1-47, 2006.
Martin Karplus. Molecular Dynamics of Biological Macromolecules: A Brief History and Perspective. Biopolymers, 68:350-358, 2003.
J. Andrew McCammon, Bruce R. Gelin, and Martin Karplus. Dynamics of Folded Proteins, Nature, 267:585-590, 1977.
Helmut Heller, Michael Schaefer, and Klaus Schulten. Molecular Dynamics Simulation of a Bilayer of 200 lipids in the Gel and in the Liquid Crystal-phases. Journal of Physical Chemistry, 97:8343-8360, 1993.
Laxmikant Kalé. Application-Oriented and Computer-Science-Centered HPCC Research. HPCC Workshop, 1994.
Mark Nelson, William Humphrey, Attila Gursoy, Andrew Dalke, Laxmikant Kalé, Robert D. Skeel, and Klaus Schulten. NAMD - A Parallel, Object-oriented Molecular Dynamics Program. International Journal of Supercomputer Applications and High Performance Computing, 10:251-268, 1996.
Marcos Sotomayor, David P. Corey, and Klaus Schulten. In Search of the Hair-cell Gating Spring: Elastic Properties of Ankyrin and Cadherin Repeats.. Structure, 13:669-682, 2005.
Marcos Sotomayor and Klaus Schulten. Single-molecule Experiments in vitro and in silico. Science, 316:1144-1148, 2007.
Fangqiang Zhu, Emad Tajkhorshid, and Klaus Schulten. Theory and Simulation of Water Permeation in Aquaporin-1. Biophysical Journal, 86:50-57, 2004.
Ly Le, Eric H. Lee, David J. Hardy, Thanh N. Truong, and Klaus Schulten. Molecular Dynamics Simulations Suggest that Electrostatic Funnel Directs Binding of Tamiflu to Influenza N1 Neuraminidases. PLoS Comput. Biol., 6:e1000939, 2010.
Peter L. Freddolino, Anton S. Arkhipov, Steven B. Larson, Alexander McPherson, and Klaus Schulten. Molecular Dynamics Simulations of the Complete Satellite Tobacco Mosaic Virus. Structure, 14:437-449, 2006.
Anton Arkhipov, Peter L. Freddolino, and Klaus Schulten. Stability and Dynamics of Virus Capsids Described by Coarse-grained Modeling. Structure, 14:1767-1777, 2006.
Appendix
“Mechanical unfolding intermediates in titin modules.”
Piotr E. Marszalek, Hui Lu, Hongbin Li, Mariano Carrion-Vazquez, Andres F. Oberhauser, Klaus Schulten, and Julio M. Fernandez. Nature, 402:100-103, 1999.
(Citations: 524)
“Control of the selectivity of the aquaporin water channel family by global orientational tuning.”
Emad Tajkhorshid, Peter Nollert, Morten Ø. Jensen, Larry J. W. Miercke, Joseph O'Connell, Robert M. Stroud, and Klaus Schulten. Science, 296:525-530, 2002.
(Citations: 497)
“Unfolding of titin immunoglobulin domains by steered molecular dynamics simulation. ”
Hui Lu, Barry Isralewitz, André Krammer, Viola Vogel, and Klaus Schulten. Biophysical Journal, 75:662-671, 1998.
(Citations: 439)
“Molecular dynamics study of unbinding of the avidin-biotin complex.”
Sergei Izrailev, Sergey Stepaniants, Manel Balsera, Yoshi Oono, and Klaus Schulten. Biophysical Journal, 72:1568-1581, 1997.
(Citations: 434)
“Steered molecular dynamics and mechanical functions of proteins.”
Barry Isralewitz, Mu Gao, and Klaus Schulten. Current Opinion in Structural Biology, 11:224-230, 2001.
(Citations: 396)
“Single-molecule experiments in vitro and in silico.”
Marcos Sotomayor and Klaus Schulten. Science, 316:1144-1148, 2007.
(Citations: 234)
“Energetics of glycerol conduction through aquaglyceroporin GlpF.”
Morten Ø. Jensen, Sanghyun Park, Emad Tajkhorshid, and Klaus Schulten. Proceedings of the National Academy of Sciences, USA, 99:6731-6736, 2002.
(Citations: 211)
“The key event in force-induced unfolding of titin's immunoglobulin domains.”
Hui Lu and Klaus Schulten. Biophysical Journal, 79:51-65, 2000.
(Citations: 209)
“Imaging alpha-hemolysin with molecular dynamics: Ionic conductance, osmotic permeability and the electrostatic potential map.”
Aleksij Aksimentiev and Klaus Schulten. Biophysical Journal, 88:3745-3761, 2005.
(Citations: 200)
“Oxygen and proton pathways in cytochrome c oxidase.”
Ivo Hofacker and Klaus Schulten. PROTEINS: Structure, Function, and Genetics, 30:100-107, 1998.
(Citations: 195)
“Sizing DNA using a nanometer-diameter pore. ”
J. B. Heng, C. Ho, T. Kim, R. Timp, A. Aksimentiev, Y. V. Grinkova, S. Sligar, K. Schulten, and G. Timp. Biophysical Journal, 87:2905-2911, 2004.
(Citations: 193)
“Forced unfolding of the fibronectin type III module reveals a tensile molecular recognition switch.”
André Krammer, Hui Lu, Barry Isralewitz, Klaus Schulten, and Viola Vogel. Proceedings of the National Academy of Sciences, USA, 96:1351-1356, 1999.
(Citations: 190)
“Microscopic kinetics of DNA translocation through synthetic nanopore.”
Aleksij Aksimentiev, Jiunn Benjamin Heng, Gregory Timp, and Klaus Schulten. Biophysical Journal, 87:2086-2097, 2004.
(Citations: 180)
“Orientation discrimination of single stranded DNA inside the α-hemolysin membrane channel. ”
Jerome Mathé, Aleksei Aksimentiev, David R. Nelson, Klaus Schulten, and Amit Meller. Proceedings of the National Academy of Sciences, USA, 102:12377-12382, 2005.
(Citations: 162)
“Steered molecular dynamics simulations of force-induced protein domain unfolding.”
Hui Lu and Klaus Schulten. PROTEINS: Structure, Function, and Genetics, 35:453-463, 1999.
(Citations: 161)


