VMD-L Mailing List
From: Marcelo Puiatti (marcelo.puiatti_at_gmail.com)
Date: Fri Apr 27 2007 - 18:12:44 CDT
- Next message: Axel Kohlmeyer: "Re: increase light under depth cueing"
- Previous message: Olaf Lenz: "Re: increase light under depth cueing"
- Next in thread: Axel Kohlmeyer: "Re: Visualization and reading problems"
- Reply: Axel Kohlmeyer: "Re: Visualization and reading problems"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ] [ attachment ]
Dear Users:
I'm a user of NAMD 2.6 and VMD 1.85.
I'm trying to minimize a protein in a periodic box of 80 x 80 x 150 A,
starting from Amber topologgies and coordinates, but in 2 of my attempts
I've found some weird things.
Firts of all, I've constrained the protein and moved only the solvent
molcules, but the solvent box was divided in exatctly 8 parts and nothing
happened to the protein. Each "scrambled" fragment separated by an empty
region passing through the geometric center of the system. However and even
though there were atoms from the solvent too close to the protein, no close
contact messages appeared!!!, more surprisingly the energies were within the
normal values!!!
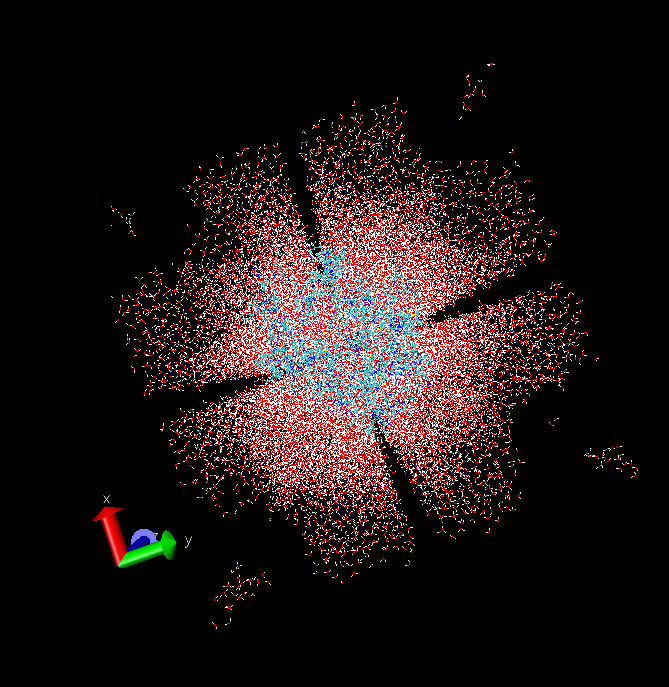
See Opt-solv.png
In the second attempt, the result was slightly different, the water box was
OK, but now the protein was scrambled divided in 8 parts. The solvent box
had a hole where the protein should be placed. Again there were no problems
with the energies, the values obtained were the same as those obtained in a
similar simulation with Amber 7.
See Min-2.png
The atoms move from one corner of the box to the other corner, with a
rotation, or a reflexion. (see file cambio delugar.pdf)
>From the blue corner to the red cornes
See cambio de lugar.pdf
We've read the coordinates with vmd using both .pdb files, .coord files
(from namd) and we've also attempted to convert .dcd files to Amber with
ptraj, but we allways obtained the same results
As initial input we've used amber files *.top and *.crd as (amber) parmfile
and ambercoor respectively.
I presume that there are reading problems between Amber format and namd or
VMD, specially with PBC, however it is quite strange that the energies do
not seem to correspond to the displayed (weird) system.
Exactly the same protocol works fine with several smaller proteins inside an
almost cubic box.
What is the right protocol to follow in order to run NAMD simulations
starting with Amber files?
Why the same protocol works well with one system and not with another?
Marcelo
P.D.
I attached the .conf file
set temperature 300
set outputname Min-2
firsttimestep 0
################################
## SIMULATION PARAMETERS ##
################################
# Input
paraTypeCharmm off
paratypeXplor off
amber on
parmfile Protein.top
ambercoor Protein.crd
temperature $temperature
# Force-Field Parameters
exclude scaled1-4
1-4scaling 0.833333 # =1/1.2, default is 1.0
cutoff 11.5
switching on
switchdist 9.
pairlistdist 13.5
# Integrator Parameters
timestep 1.5 ;# 2fs/step
rigidBonds all ;# needed for 2fs steps
nonbondedFreq 1
fullElectFrequency 2
stepspercycle 20
# Constant Temperature Control
langevin on ;# do langevin dynamics
langevinDamping 5 ;# damping coefficient (gamma) of 5/ps
langevinTemp $temperature
langevinHydrogen off ;# don't couple langevin bath to hydrogens
# Periodic Boundary Conditions
cellBasisVector1 80.15 0. 0.
cellBasisVector2 0. 79.24 0.
cellBasisVector3 0. 0.0 150.64
cellOrigin 45.52 45.94 78.17
wrapAll on
# PME (for full-system periodic electrostatics)
PME yes
PMEGridSizeX 144
PMEGridSizeY 144
PMEGridSizeZ 256
# Constant Pressure Control (variable volume)
useGroupPressure yes ;# needed for rigidBonds
useFlexibleCell no
useConstantArea no
# BerendsenPressure on
# BerendsenPressureTarget 1.01325
# BerendsenPressureCompressibility 4.57E-5
# BerendsenPressureRelaxationTime 1000
# BerendsenPressureFreq 20 #chequear
# langevinPiston on
# langevinPistonTarget 1.01325 ;# in bar -> 1 atm
# langevinPistonPeriod 100.
# langevinPistonDecay 50.
# langevinPistonTemp $temperature
#Constraints
constraints on
consexp 6
consRef protein.pdb
consKFile protein-constraint.pdb
consKCol B
# Output
outputName $outputname
restartfreq 200 ;# 500steps = every 1ps
dcdfreq 500
xstFreq 500
outputEnergies 50
outputPressure 50
#############################################################
## EXECUTION SCRIPT ##
#############################################################
# Minimization
minimize 5000

- application/pdf attachment: cambio_de_lugar.pdf
- Next message: Axel Kohlmeyer: "Re: increase light under depth cueing"
- Previous message: Olaf Lenz: "Re: increase light under depth cueing"
- Next in thread: Axel Kohlmeyer: "Re: Visualization and reading problems"
- Reply: Axel Kohlmeyer: "Re: Visualization and reading problems"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ] [ attachment ]


