VMD-L Mailing List
From: Kailee (kaileeamber_at_googlemail.com)
Date: Thu Mar 22 2007 - 10:04:44 CDT
- Next message: John Stone: "Re: How to superimpose trajectories"
- Previous message: John Stone: "Re: colors"
- In reply to: John Stone: "Re: How to superimpose trajectories"
- Next in thread: John Stone: "Re: How to superimpose trajectories"
- Reply: John Stone: "Re: How to superimpose trajectories"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ] [ attachment ]
Dear John,
Thanks very much for your reply.
The extra atoms I got is because of the selection is not updated as the
other frames are drawn, and after I ticked 'update selection every frame',
the problem has gone.
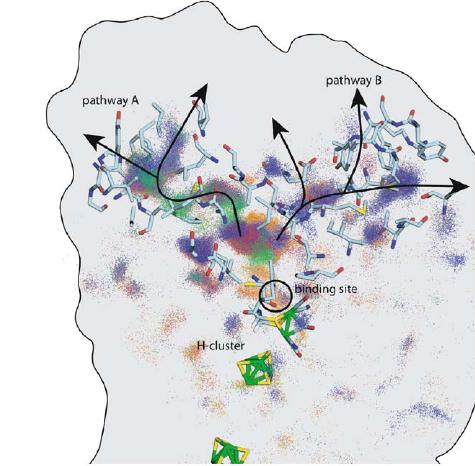
And the picture I mentioned in my last email, I have attached again. The one
named 'example' is the picture I want to get, and the 'snap' is the one I
can currently make myself. Can you give me some advice on how to improve it
please?
Thank you very much for your great help.
Best regards,
Kailee
On 3/21/07, John Stone <johns_at_ks.uiuc.edu> wrote:
>
>
> Kailee,
> I went back and looked to see the picture attachment you referred to
> but didn't find one in the email you sent. Let us know if you need
> more assistance.
>
> John Stone
> vmd_at_ks.uiuc.edu
>
> On Wed, Mar 21, 2007 at 03:26:20PM -0500, John Stone wrote:
> >
> > Hi,
> >
> > On Wed, Mar 21, 2007 at 02:14:33PM +0000, Kailee wrote:
> > [...]
> > > 1) I tried to superimpose the hydrogen molecule trajectories, what I
> did
> > > was: select the representation of hydrogen molecule, and in the
> > > 'trajectory-> draw multiple frames', I changed 'now' to '0:', is it
> the
> > > right way to do this?
> >
> > You'll need to have an end frame, so if you had 10 frames, you'd use
> "0:9"
> >
> > > 2) Because I don't want to look at all the hydrogens but only
> hydrogens
> > > within a certain distance of the protein center, so in the
> 'selections', I
> > > typed 'resname H2 and (within 50 of name FE)', as I want to look
> at the
> > > hydrogen molecules within 50A of the Fe atom. However, it turned out
> that
> > > some hydrogens which are very obvious not within 50A of Fe still
> there.
> >
> > When you use the draw multiple frames feature, the selection is not
> updated
> > as the other frames are drawn, the same selection is used to draw all of
> > the "multiple frames", so this could explain why you're getting extra
> > atoms shown. Try showing just the "now" frame and see if only the
> > expected atoms are shown. The other possibility is PBC as you mentioned
> > below:
> >
> > > I am
> > > thinking it might because of the periodic boundary problem that some
> > > hydrogens flied out of the primary box and I should reimage them back
> to the
> > > central box. Is it right? And if so, how can I do it in VMD?
> >
> > You could try using the PBC tools scripts (plugin in VMD 1.8.6) to
> > do this for you.
> >
> > > 3) And also when I superimposed the hydrogen trajecotries, the
> > > representation of the protein residues became really not clear, I
> tried
> > > different draw styles but still can't find out how to do it.
> >
> > Can you be specific as to "really not clear" means? Do you have a
> screen
> > shot that shows what you're getting?
> >
> > When you use the "draw multiple timesteps" feature, it is often useful
> > to set the coloring method to "Timestep" so that you can differentiate
> > them.
> >
> > > Actually I have
> > > attached a picture that I copied from a paper, what I want to do is
> almost
> > > exactly what he did, but just using different protein molecule, can
> anyone
> > > who is familiar with VMD guide me how to make a picture like that
> please?
> >
> > Let me look at it and get back to you.
> >
> > John Stone
> > vmd_at_ks.uiuc.edu
> >
> > --
> > NIH Resource for Macromolecular Modeling and Bioinformatics
> > Beckman Institute for Advanced Science and Technology
> > University of Illinois, 405 N. Mathews Ave, Urbana, IL 61801
> > Email: johns_at_ks.uiuc.edu Phone: 217-244-3349
> > WWW: http://www.ks.uiuc.edu/~johns/ Fax: 217-244-6078
>
> --
> NIH Resource for Macromolecular Modeling and Bioinformatics
> Beckman Institute for Advanced Science and Technology
> University of Illinois, 405 N. Mathews Ave, Urbana, IL 61801
> Email: johns_at_ks.uiuc.edu Phone: 217-244-3349
> WWW: http://www.ks.uiuc.edu/~johns/ Fax: 217-244-6078
>

- Next message: John Stone: "Re: How to superimpose trajectories"
- Previous message: John Stone: "Re: colors"
- In reply to: John Stone: "Re: How to superimpose trajectories"
- Next in thread: John Stone: "Re: How to superimpose trajectories"
- Reply: John Stone: "Re: How to superimpose trajectories"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ] [ attachment ]


