VMD-L Mailing List
From: Diego Gomes (diego.enry_at_gmail.com)
Date: Thu Nov 02 2023 - 12:21:42 CDT
- Next message: Diego Gomes: "Re: STRIDE breaks after 100,000 protein atoms"
- Previous message: Vasista: "STRIDE breaks after 100,000 protein atoms"
- In reply to: Jirui Yang: "Re: Molefacture 2 does not generate angles & dihedrals"
- Next in thread: Pawel Kedzierski: "Re: Molefacture 2 does not generate angles & dihedrals"
- Reply: Pawel Kedzierski: "Re: Molefacture 2 does not generate angles & dihedrals"
- Reply: Pawel Kedzierski: "Re: Molefacture 2 does not generate angles & dihedrals"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ] [ attachment ]
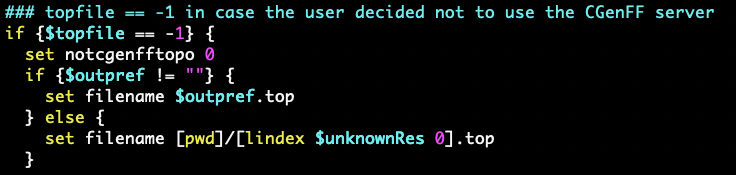
Thanks to Yupeng & Hale the issue has been found and addressed in the CVS
source.
To fix it in your installed version, please change line 2692 of
*molefacture_internals.tcl *
from
*set notcgenfftopo 1*
to
*set notcgenfftopo 0*
(see attached image)
You may find molefacture_internals.tcl at
MacOS
/Applications/VMD\
1.9.4a57-arm64-Rev12.app/Contents/vmd/plugins/noarch/tcl/molefacture2.0/molefacture_internals.tcl
Linux
vmd_install_path
/lib/vmd/plugins/noarch/tcl/molefacture2.0/molefacture_internals.tcl
Please let us know if you find any other issues.
Friendly regards,
Diego.
[image: Screenshot 2023-11-02 at 12.12.32 PM.png]
On Mon, Oct 30, 2023 at 12:58 PM Jirui Yang <jirui_at_hawaii.edu> wrote:
> I still need to regenerate your problem. However, in my most recent
> research using VMD, it has appeared that some bonds will disappear after
> running calculations. We thought it was a bug at first. Please keep
> me updated on this issue.
> Respectfully
> Jirui
>
> On Mon, Oct 30, 2023 at 3:07 AM Pawel Kedzierski <
> pawel.kedzierski_at_pwr.edu.pl> wrote:
>
>> My apology for the double posting, but I've just realized that my vmd
>> was not the most recent one and I have confirmed, that the problem
>> persists in 1.9.4a57 for Linux
>> Also, at the time of saving PSF and PDB files from Molefacture I
>> answered "No" to the question, whether to download the topology from the
>> CGenFF server.
>> With regards,
>> Paweł Kędzierski
>>
>> W dniu 30.10.2023 o 08:54, Paweł Kędzierski pisze:
>> > Dear All,
>> >
>> > Since several years I use Molefacture & FFTK on my molecular modeling
>> > classes. This year I have switched to using Molefacture 2 and I have
>> > observed, that the angles and dihedrals are not generated from the
>> > bonds and therefore missing in the saved the PSF file. Perhaps I am
>> > missing something, but I don't see any option to generate them on
>> > request. On the other hand, Molefacture 1.0 available with vmd1.9.4a8
>> > saved the PSF file with complete topology information.
>> >
>> > To reproduce the problem, you may save the example below as a
>> > h2o2.mol2 file, load it into VMD, open
>> > Extensions->Modeling->Molefacture, create a new working molecule in
>> > Molefacture using File->New molecule->From selection (using the
>> > selection "all"), and then save with File->Write PSF and PDB files. In
>> > the PSF file, there are 0 angles and 0 dihedrals. The same happens
>> > with the option Build->Topology & Parameters->Export to FFTK.
>> > This was tested using vmd1.9.4a55 on Linux Debian 12.
>> > How can I fix this, to create the PSF file with complete topology,
>> > usable with FFTK?
>> > With regards,
>> > Paweł Kędzierski
>> >
>> > ----<h2o2.mol2 file contents----
>> > @<TRIPOS>MOLECULE
>> > *****
>> > 4 3 0 0 0
>> > SMALL
>> > GASTEIGER
>> >
>> > @<TRIPOS>ATOM
>> > 1 O 0.1046 -0.1116 -0.5641 O.3 1 UNL1
>> > -0.2528
>> > 2 O 0.5297 -0.9231 0.5641 O.3 1 UNL1
>> > -0.2528
>> > 3 H -0.6326 0.3369 -0.1076 H 1 UNL1
>> > 0.2528
>> > 4 H -0.0658 -1.6768 0.3903 H 1 UNL1
>> > 0.2528
>> > @<TRIPOS>BOND
>> > 1 1 2 1
>> > 2 1 3 1
>> > 3 2 4 1
>> > ----<cut above this line>-----
>> >
>> >
>>
>
-- Diego Enry B. Gomes, PhD Department of Physics at Auburn University & NIH Center for Macromolecular Modeling and Visualization Leach Science Center - Ste. 3182 - Auburn, AL dgomes_at_auburn.edu
- Next message: Diego Gomes: "Re: STRIDE breaks after 100,000 protein atoms"
- Previous message: Vasista: "STRIDE breaks after 100,000 protein atoms"
- In reply to: Jirui Yang: "Re: Molefacture 2 does not generate angles & dihedrals"
- Next in thread: Pawel Kedzierski: "Re: Molefacture 2 does not generate angles & dihedrals"
- Reply: Pawel Kedzierski: "Re: Molefacture 2 does not generate angles & dihedrals"
- Reply: Pawel Kedzierski: "Re: Molefacture 2 does not generate angles & dihedrals"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ] [ attachment ]


