NAMD 3.0 Alpha, GPU-Resident Single-Node-Per-Replicate Test Builds
This page contains special developmental early "alpha" test builds of NAMD 3.0 intended to provide very early testing access to the NAMD user and developer communities.
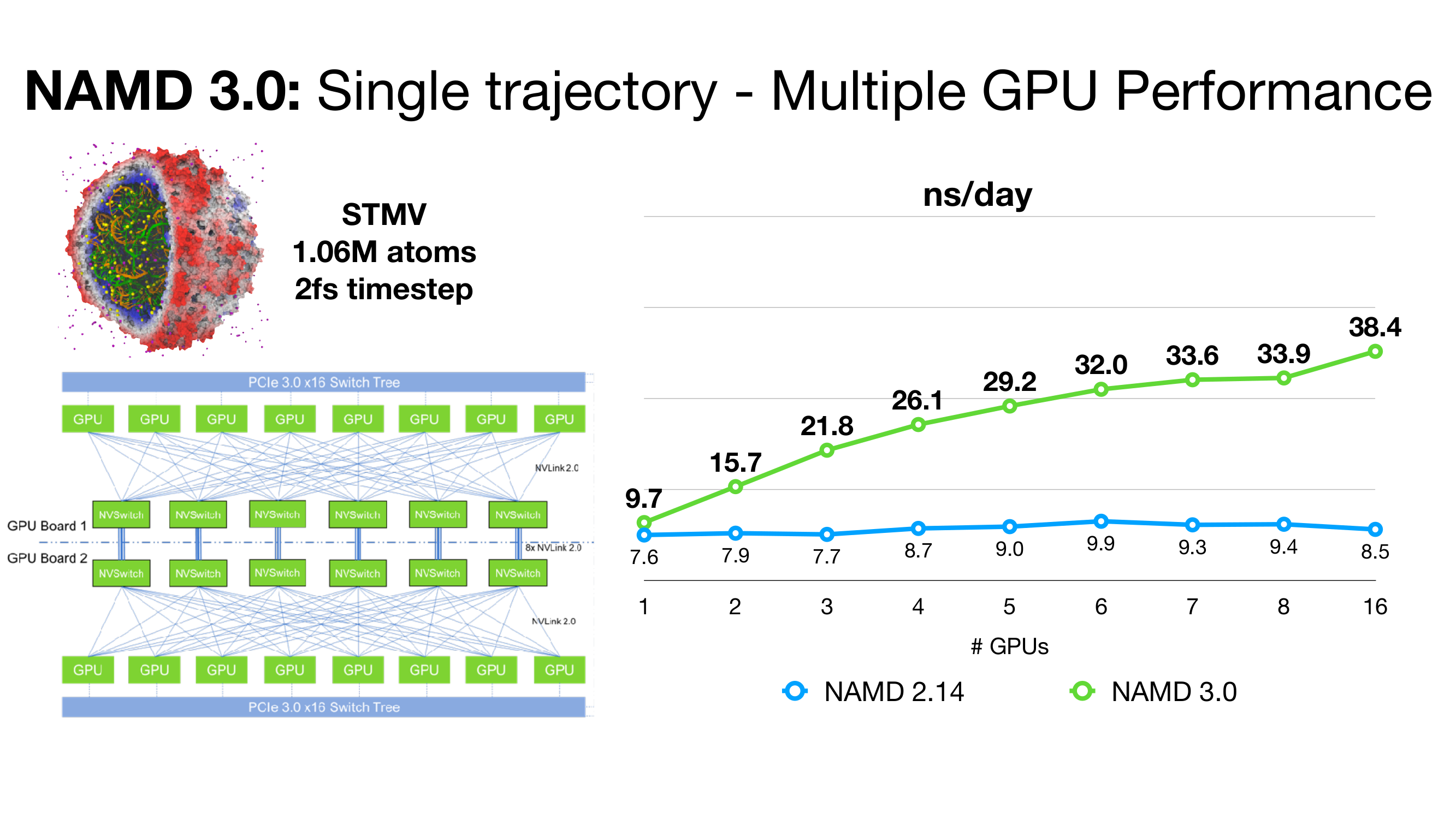
on an NVIDIA DGX-2 w/ Tesla V100 GPUs.
GPU acceleration feature provides up to 2x the
performance of prior versions for appropriate MD
simulations on modern GPUs.
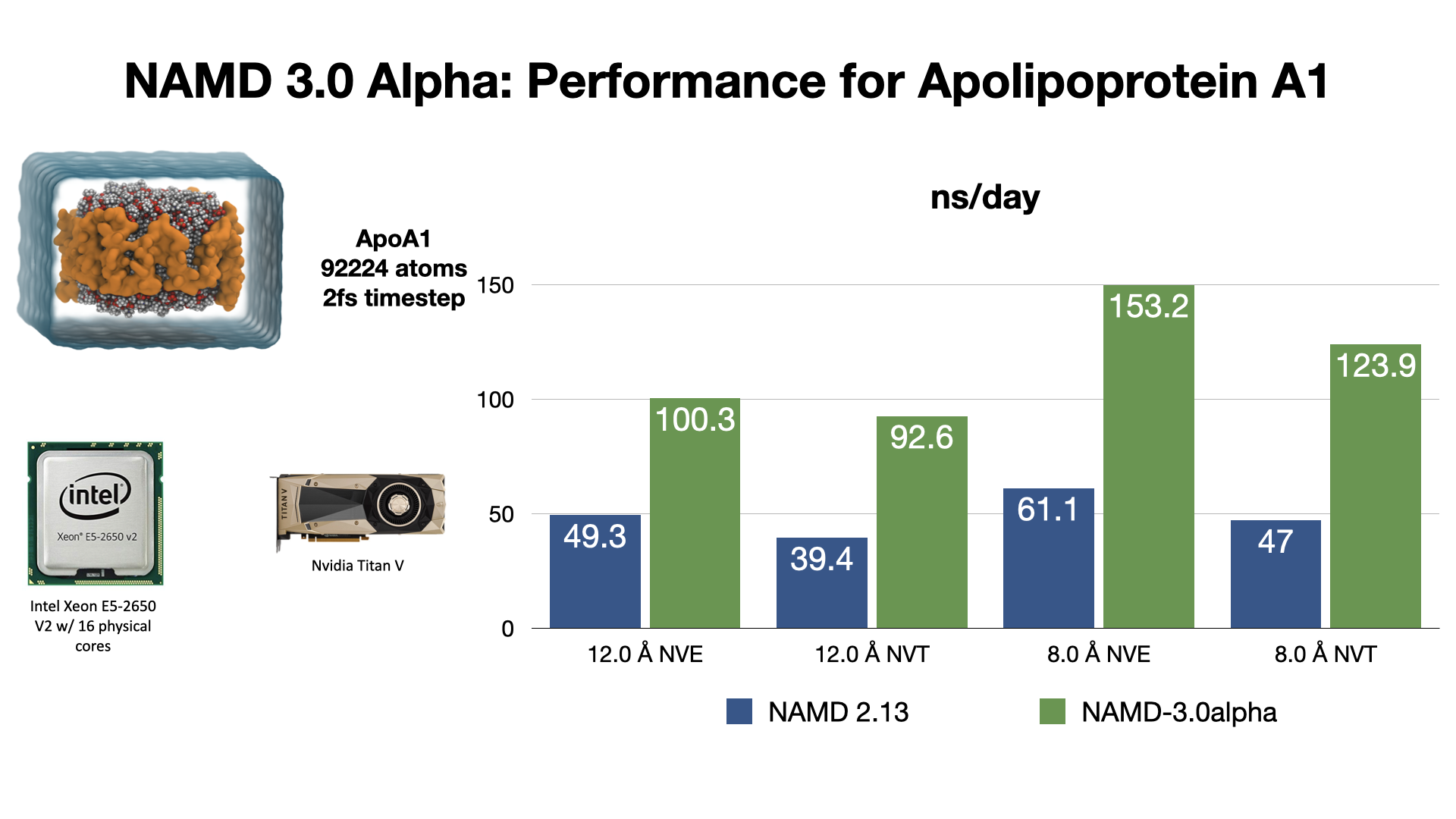
An exciting NAMD 3.0 feature being developed is a GPU-resident single-node-per-replicate computation mode that can speed up simulations up to two or more times for modern GPU architectures, e.g., Volta, Turing, Ampere.
This new NAMD simulation mode maximizes performance of small- to moderate-sized molecular dynamics simulations suitable for the computational capabilities of a single GPU-accelerated compute node, by virtue of new GPU-specific streamlined code paths that offload the integrator and rigid bond constraints to the GPU, while bypassing and thereby eliminating CPU activities and associated overheads that would otherwise slow down these simulations. With current developmental test builds of NAMD 3.0 alpha, the new code paths can provide a 2x performance gain compared to previous versions of NAMD running the same simulation on the same hardware.
What MD Simulations Are Well-Suited to NAMD 3.0 Alpha Versions?
This scheme is intended for small to medium systems (10 thousand to 1 million atoms). For larger simulations, you should stick to the regular integration scheme, e.g., as used in NAMD 2.x.
This scheme is intended for modern GPUs, and it might slow your simulation down if you are not running on a Volta, Turing, or Ampere GPU! If your GPU is older, we recommend that you stick to NAMD 2.x.
The single-node version of NAMD 3.0 has almost everything offloaded to the GPU, so large CPU core counts are NOT necessary to get good performance. We recommend running NAMD with a low +p count, maybe 2-4 depending on system size, especially if the user plans on running multiple replica simulations within a node.
NAMD 3.0 Feature Notes:
NAMD contains a lot of code! We currently don't have corresponding GPU code for everything that the original CPU is capable of handling. The consequence is that some features are still not supported, and others are supported but don't gain much performance (yet). Most of the supported features are related to equilibrium simulations, but some biasing schemes are upcoming.
The essential feature support includes:
- Constant energy
- Constant temperature with Langevin damping
- Constant temperature with stochastic velocity rescaling
- Constant pressure with Langevin piston
- Particle-mesh Ewald (PME) for long-range electrostatics
- Rigid bond constraints
- Multiple time stepping
The advanced feature support includes:
- Multi-copy simulation (e.g. replica-exchange)
- External electric field
- Harmonic restraints
- Steered molecular dynamics (SMD) - alpha 12
- Group position restraints - alpha 13
NAMD 3.0 Simulation Parameter Changes:
Adding the following keyword to your NAMD configuration file enables the single-node GPU-resident integration scheme required for high performance:
- CUDASOAintegrate on
To attain good performance, it is very important to set the margin performance tuning parameters. (defaulting in NAMD 2.x to 0). If margin is left UNDEFINED in the simulation configuration file with CUDASOAintegrate enabled, NAMD will automatically set it as follows:
- margin 4
Beginning with NAMD 3.0 alpha 4, it is no longer necessary to set stepsPerCycle or pairlistsPerCycle, as NAMD 3.0 alpha4 and later versions automatically adjust these values on-the-fly at runtime. (defaulting in NAMD 2.x to 20, 2, respectively). In addition, frequent output will limit performance. Most importantly, outputEnergies (defaulting in NAMD 2.x to 1) should be set much higher, e.g., 400.
Notes on minimization with NAMD 3.0:
Due to the way minimization algorithms are implemented in NAMD (primarily CPU-based) and the way that NAMD performas spatial decomposition of the molecular system into patches, minimization and dynamics should be performed in two separate NAMD runs. This ensures that NAMD will recompute the patch decomposition for dynamics optimally, yielding the best overall performance for the production part of the simulation. Beyond this the CUDASOAintegrate parameter must be set to off, to allow the CPU minimization code path to be used correctly, as shown below:
- CUDASOAintegrate off
Notes on NAMD 3.0 Group Position Restraints
Exclusive (for now) to the single-node GPU-resident version is the new group position restraints advanced feature. It allows the user to define a restraint either between the centers-of-mass of two groups of atoms or between the center-of-mass of a single group of atoms and a fixed reference point.
The following example shows a distanceZ collective variable
in a membrane bilayer equilibration simulation and how you can use
group position retraints to achieve the same objective.
##### COLVARS #####
colvar {
name popc_head_upper
distanceZ {
ref {
dummyAtom ( 0.000, 0.000, 0.000 )
}
main {
atomsFile restraints/popc_head_upper.ref
atomsCol B
atomsColValue 1.0
}
axis (0, 0, 1)
}
}
harmonic {
colvars popc_head_upper
centers 19
forceConstant 5
}
colvar {
name popc_head_lower
distanceZ {
ref {
dummyAtom ( 0.000, 0.000, 0.000 )
}
main {
atomsFile restraints/popc_head_lower.ref
atomsCol B
atomsColValue 1.0
}
axis (0, 0, 1)
}
}
harmonic {
colvars popc_head_lower
centers -19
forceConstant 5
}
##### NAMD 3 GROUP RESTRAINTS #####
groupRestraints on
group1RefPos popc_upper 0 0 0
group2File popc_upper restraints/popc_head_upper.txt ;# atom indices for POPC head groups
groupResExp popc_upper 2
groupResK popc_upper 2.5
groupResCenter popc_upper 0 0 19
groupResX popc_upper off
groupResY popc_upper off
groupResZ popc_upper on
group1RefPos popc_lower 0 0 0
group2File popc_lower restraints/popc_head_lower.txt ;# atom indices for POPC head groups
groupResExp popc_lower 2
groupResK popc_lower 2.5
groupResCenter popc_lower 0 0 -19
groupResX popc_lower off
groupResY popc_lower off
groupResZ popc_lower on
More information is available in the updated NAMD 3.0 User Guide, available in PDF and HTML formats. The new group position restraints feature with all of its config file options is documented here.
Notes on NAMD 3.0 Multi-GPU Scaling Per-Replicate
NAMD 3.0 alpha builds now support multi-GPU scaling of single-replicate simulations within a single node, for sets of NVLink-connected GPUs. NAMD 3.0 will drive them in GPU-resident mode, with minimal need for CPU power. In order to scale a single simulation across multiple GPUs, all pairs of devices must be "peered" with each other, either having a direct NVLink cable connection or all plugged into an NVSwitch, as is the case with the DGX series hardware. You can verify the connectivity of your GPUs by running the command "nvidia-smi topo -m" and checking that the connectivity matrix shows an "NV" connection between each pair of GPUs.
Multiple GPU devices are specified with the +devices. flag. The original support provided in the alpha 9 release requires exactly one CPU core per GPU device, meaning that the +pN CPU core count must exactly match the GPU device count specified with +devices. That restriction has been lifted since alpha 10 and later releases. Using more than one CPU core per device helps improve the performance of parts that remain on the CPU, in particular, atom migration.
NAMD 3 currently delegates one device to perform long-range electrostatics with PME, when enabled. You can specify which GPU device PME will run on by including +pmedevice deviceid in your command-line run. One issue with multi-GPU scaling is that PME causes a bottleneck when all of the non-PME work is equally divided between devices. Since alpha 10, we provide a way to control the amount of work assigned to the PME device by scaling back the number of PEs (CPU cores) for that device using the +pmePEs command line parameter. This is best shown through the example of scaling an STMV-sized system (around 1M atoms) on DGX-A100:
./namd3 +p57 +pmePEs 1 +setcpuaffinity +devices 0,1,2,3,4,5,6,7 myconf.namdIn this example, 8 PEs are used per device, except for the PME device which is limited to 1, providing a balanced load for each device. Notice that the total number of PEs is adjusted to (7*8 + 1) = 57.
Since alpha 11, the short-range non-bonded forces kernel can be sped up by direct calculation of force interactions rather than table lookup for non-PME steps, as might happen when using multiple time stepping. You can request direct calculation by the config file setting:
- CUDAForceTable off
The following advanced features are not yet supported by multi-GPU runs: group position restraints, steered molecular dynamics (SMD), harmonic restraints, and electric field.
The code is still evolving, and test builds will be updated frequently. Stay tuned!
NAMD 3.0 Single- and Multi-GPU-Per-Replicate Alpha Versions For Download:
These are the first builds that support both single- and multi-GPU-per replicate runs in the same binary.- NAMD_3.0alpha13_Linux-x86_64-multicore-CUDA-SingleNode.tar.gz (Standard simulation.) (July 24, 2022)
- NAMD_3.0alpha13_Linux-x86_64-netlrts-smp-CUDA-SingleNode.tar.gz (Multi-copy GPU simulations. For multi-GPU machines use: "+devicesperreplica 1") (July 24, 2022)
- NAMD_3.0alpha12_Linux-x86_64-multicore-CUDA-SingleNode.tar.gz (Standard simulation.) (June 22, 2022)
- NAMD_3.0alpha12_Linux-x86_64-netlrts-smp-CUDA-SingleNode.tar.gz (Multi-copy GPU simulations. For multi-GPU machines use: "+devicesperreplica 1") (June 22, 2022)
- NAMD_3.0alpha11_Linux-x86_64-multicore-CUDA-SingleNode.tar.gz (Standard simulation.) (May 24, 2022)
- NAMD_3.0alpha11_Linux-x86_64-netlrts-smp-CUDA-SingleNode.tar.gz (Multi-copy GPU simulations. For multi-GPU machines use: "+devicesperreplica 1") (May 24, 2022)
- NAMD_3.0alpha10_Linux-x86_64-multicore-CUDA-SingleNode.tar.gz (Standard simulation.) (March 24, 2022)
- NAMD_3.0alpha10_Linux-x86_64-netlrts-smp-CUDA-SingleNode.tar.gz (Multi-copy GPU simulations. For multi-GPU machines use: "+devicesperreplica 1") (March 24, 2022)
- NAMD_3.0alpha9_Linux-x86_64-multicore-CUDA-SingleNode.tar.gz (Standard simulation.) (February 28, 2021)
- NAMD_3.0alpha9_Linux-x86_64-netlrts-smp-CUDA-SingleNode.tar.gz (Multi-copy GPU simulations. For multi-GPU machines use: "+devicesperreplica 1") (February 28, 2021)
NAMD 3.0 Single-GPU-Per-Replicate Alpha Versions For Download:
The builds in this section support only a SINGLE-GPU offloading scheme. Builds for multi-GPU scaling per-replicate are posted in the section above.- NAMD_3.0alpha8_Linux-x86_64-multicore-CUDA-SingleNode.tar.gz (Standard simulation.) (December 21, 2020)
- NAMD_3.0alpha8_Linux-x86_64-netlrts-smp-CUDA-SingleNode.tar.gz (Multi-copy GPU simulations. For multi-GPU machines use: "+devicesperreplica 1") (December 21, 2020)
- NAMD_3.0alpha7_Linux-x86_64-multicore-CUDA-SingleNode.tar.gz (Standard simulation.) (October 16, 2020)
- NAMD_3.0alpha7_Linux-x86_64-netlrts-smp-CUDA-SingleNode.tar.gz (Multi-copy GPU simulations. For multi-GPU machines use: "+devicesperreplica 1") (October 16, 2020)
- NAMD_3.0alpha6_Linux-x86_64-multicore-CUDA-SingleNode.tar.gz (Standard simulation.) (August 12, 2020)
- NAMD_3.0alpha6_Linux-x86_64-netlrts-smp-CUDA-SingleNode.tar.gz (Multi-copy GPU simulations. For multi-GPU machines use: "+devicesperreplica 1") (August 12, 2020)
- NAMD_3.0alpha5_Linux-x86_64-multicore-CUDA-SingleNode.tar.gz (Standard simulation.) (July 22, 2020)
- NAMD_3.0alpha5_Linux-x86_64-netlrts-smp-CUDA-SingleNode.tar.gz (Multi-copy GPU simulations. For multi-GPU machines use: "+devicesperreplica 1") (July 22, 2020)
- NAMD_3.0alpha4_Linux-x86_64-multicore-CUDA-SingleNode.tar.gz (Standard simulation.) (July 18, 2020)
- NAMD_3.0alpha4_Linux-x86_64-netlrts-smp-CUDA-SingleNode.tar.gz (Multi-copy GPU simulations. For multi-GPU machines use: "+devicesperreplica 1") (July 18, 2020)
- NAMD_3.0alpha3_Linux-x86_64-multicore-CUDA-SingleNode.tar.gz (Standard simulation.) (June 24, 2020)
- NAMD_3.0alpha3_Linux-x86_64-netlrts-smp-CUDA-SingleNode.tar.gz (Multi-copy GPU simulations. For multi-GPU machines use: "+devicesperreplica 1") (June 24, 2020)
- NAMD_3.0alpha2_Linux-x86_64-multicore-CUDA-SingleNode.tar.gz (Standard simulation.) (June 11, 2020)
- NAMD_3.0alpha2_Linux-x86_64-netlrts-smp-CUDA-SingleNode.tar.gz (Multi-copy GPU simulations. For multi-GPU machines use: "+devicesperreplica 1") (June 11, 2020)
- NAMD_3.0alpha_Linux-x86_64-multicore-CUDA-SingleNode.tar.gz (Standard simulation.) (May 11, 2020)
- NAMD_3.0alpha_Linux-x86_64-netlrts-smp-CUDA-SingleNode.tar.gz (Multi-copy GPU simulations. For multi-GPU machines use: "+devicesperreplica 1") (May 11, 2020)
For more information about NAMD and support inquiries:
For general NAMD information, see the main NAMD home page http://www.ks.uiuc.edu/Research/namd/
For your convenience, NAMD has been ported to and will be installed on the machines at the NSF-sponsored national supercomputing centers. If you are planning substantial simulation work of an academic nature you should apply for these resources. Benchmarks for your proposal are available at http://www.ks.uiuc.edu/Research/namd/performance.html
The Theoretical and Computational Biophysics Group encourages NAMD users to be closely involved in the development process through reporting bugs, contributing fixes, periodical surveys and via other means. Questions or comments may be directed to namd@ks.uiuc.edu.
We are eager to hear from you, and thank you for using our software!


