QwikMD - Easy and Fast Molecular Dynamics
Run Tab
Selection Window:
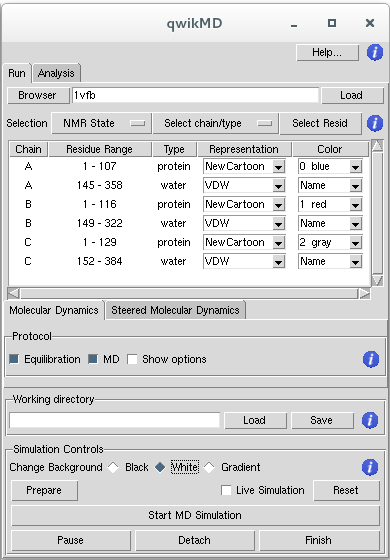
The selection section of QwikMD allows the user to select each parts of the PDB will be prepared for the MD simulation. For example, structures obtained with NMR spectroscopy usually have more than one state of the protein. It is also common that PDB structures solved with X-ray Crystallography have oligomers of the protein, separated in different chains. These oligomers are frequently an effect of the crystallization process and the protein in solution is presented as a monomer. QwikMD allows the user to select the desired NMR state or protein chains for the MD step.
NMR Structures
Nuclear magnetic resonance spectroscopy of proteins, usually abbreviated protein NMR, is a field of structural biology in which NMR spectroscopy is used to obtain information of the structure and dynamics of proteins, nucleic acids, and their complexes. NMR structure in the PDB usually have multiple steps. In order to start a MD simulation one have to select one of the steps as the initial coordinates. It is usual, when running more than one simulation of the same system, to select different initial steps to improve sampling of conformational structure.
Select chain/type
With this button the user can select or deselect chains and types of molecules inside these chains. The selected groups will be the one that will be present in the MD simulation prepared with QwikMD.
Select Resid
This button will open a new window where the user can do mutations, rename molecules that have wrong names change protonation states, delete parts of the molecules and also inspect the structure with a interactive residue/molecule list. Select Resid is especially important in cases where one of the molecules/ions have wrong names, or names that are different from the name used in the CHARMM force field. For instance, it is common in the PDB that Ca2+ ions have the name CA. CHARMM recognize the name CA as alpha-Carbon of protein structures, and CA resname - residue name - is not recognized by CHARMM. Select Resid allow for the user to rename CA ions to proper Calcium parameters that will be compatible with the CHARMM force field.
QwikMD Main Window
In the main window of QwikMD, every chain is separated by type of molecule forming a group - chain/type. VMD has several types of molecules, including: protein, nucleic, lipids, water, among others. The user can select different Representations for each of the groups and also different colors.
QwikMD Output Files
To run molecular dynamics simulations with NAMD at least four files are required: a Protein Data Bank (pdb) file a Protein Structure File (psf), a force field parameter file and a configuration file. During the preparation steps, where the system might be solvated, ionized, among other procedures, several files are created. QwikMD separates the files created in the preparation step in a SETUP folder, while files needed to run the MD simulations are in a RUN folder. These two folders are created inside the folder defined by the user in working directory window. With the same name as the folder created by the user, a file with .QwikMD extension allows the user to load simulations performed with QwikMD and also previously created preparation steps, like amino acid residues mutations or salt concentration.
To run a simulation prepared with QwikMD in a computer cluster or supercomputer, one needs to copy only the RUN folder.
To learn more about MD simulations check the list of reviews published by our group.
Simulation Controls
QwikMD helps the user to prepare NAMD input files to run in a range of computers, from the largest supercomputers with high-end parallel platforms to the smallest laptop computers. QwikMD also allow, through the Interactive Molecular Dynamics interface of NAMD, to run live simulations where the user can look and analyze the trajectories while they are being created.
Preparing simulations with QwikMD
When the Prepare button is pressed, scripts to perform all the steps required by the settings selected by user. Two folders will be created in the Working directory: files created in the preparation step in a SETUP folder, while files needed to run the MD simulations are in a RUN folder.
Running NAMD Live
If you want to run Live Simulation, make sure you have the corresponding box checked before you click Prepare and then just press Start for a live simulation.
WARNING:
Always click to FINISH your simulation before leaving VMD or closing QwikMD window. NAMD activities started from VMD will continue to run in background for all the steps requested unless you make sure the simulation was aborted before leaving QwikMD.
Running NAMD in a Supercomputer
To run NAMD in a supercomputer copy the RUN folder to your folder in the supercomputer file system and run NAMD following the procedures for the specific computer.
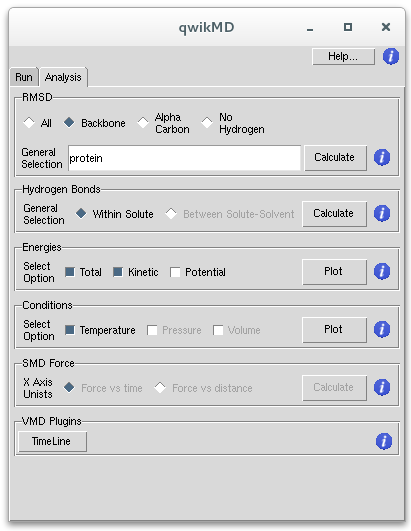
Analysis Tab
VMD is a powerful tool for analysis of structures and trajectories and should be used as a tool to think. Numerous tools for analysis are available under the VMD Main menu item Extensions - Analysis. In addition to these built-in tools, VMD users often use custom-written scripts to analyze desired properties of the simulated systems. VMD Tcl scripting capabilities are very extensive, and provide boundless opportunities for analysis. QwikMD provides the user with some of the most employed analysis tools, allowing also the analysis while performing live NAMD sections.
|
Analysis Available within QwikMD
|
|


