VMD-L Mailing List
From: Mohan maruthi sena (maruthi.sena_at_gmail.com)
Date: Thu Mar 31 2016 - 22:55:00 CDT
- Next message: John Stone: "Re: Trouble launching VMD 1.9.2 from the dock in Mac OS X el capitan"
- Previous message: Ky Wildermuth: "issue drawing a PBC box centered on a selection"
- In reply to: Mayne, Christopher G: "Re: FFTK dihedral fitting"
- Next in thread: Mayne, Christopher G: "Re: Re: FFTK dihedral fitting"
- Reply: Mayne, Christopher G: "Re: Re: FFTK dihedral fitting"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ] [ attachment ]
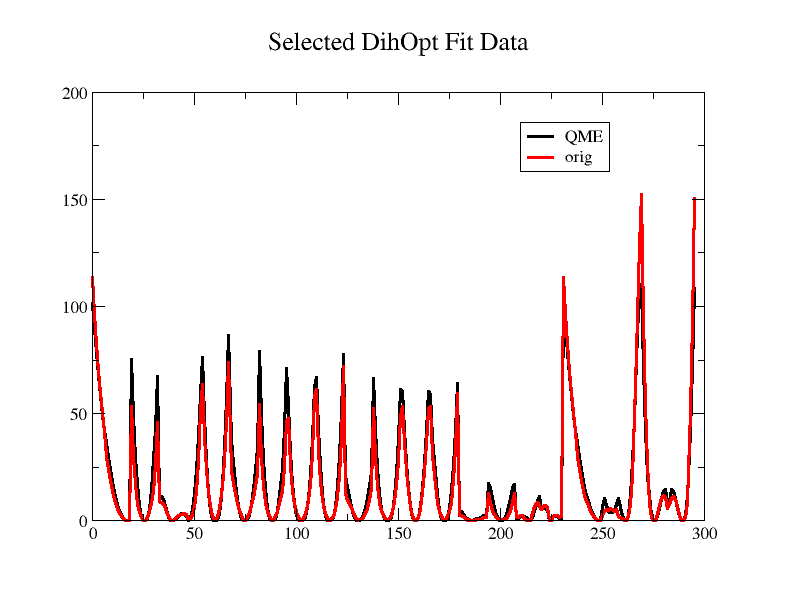
Thank you, sir, for a reply. I will try with VMD 1.9.3 alpha release and
try for a better fit. Please find the attached plot with y-axis visible.
Can you please comment on it.
Thanks & Regards,
Mohan
On Thu, Mar 31, 2016 at 8:35 PM, Mayne, Christopher G <cmayne2_at_illinois.edu>
wrote:
> Mohan,
>
> By in large, your fit looks quite good. Without the y-axis, it’s a little
> hard to tell how much the inconsistencies matter. You shouldn’t expect the
> MM PES to match the QM *exactly*, and depending on the complexity of your
> molecular system, might actually be a sign of overfitting. The only part
> the concerns me a bit is the second to last set of peaks, in which the
> shape appears to be different.
>
> In response to your specific questions:
>
> 1) Yes! I came across this same problem and built a module that allows
> you to load in the Gaussian log files, which are parsed for both the energy
> and the coordinates. The plugin then shows an interactive plot that is
> color coded by the dihedral scan. As you move through the coordinates in
> the VMD window, and indicator will show you where you are on the PES, and
> the dihedral that was scanned is shown in a color-coded licorice
> representation. This allows you to analyze the QM PES with respect to the
> structure.
>
> This tools is in the 1.9.3 alpha releases, under the Scan Torsion tab just
> below the Gaussian Settings via the “Open Torsion Explorer” button.
>
> 2) By default ffTK excludes torsions terminating in hydrogens from the
> scan. If you want to explicitly scan these torsions, e.g., for polar
> hydrogens, then you can add them to the “Dihedrals to Scan” box. During
> the fitting, however, all dihedrals provided in the “Dihedral Parameter
> Settings” will be fit, and should include dihedrals that terminate with
> hydrogens.
>
>
> Regards,
> Christopher Mayne
>
> > On Mar 30, 2016, at 9:37 AM, Mohan maruthi sena <maruthi.sena_at_gmail.com>
> wrote:
> >
> > Hi all,
> > I am trying to generate force field parameters for a molecule
> which contains 41 atoms. Except dihedral parameters, I am able to generate
> charges, bonded parameters. I am trying to fit dihedrals and tried to
> refine it but still the fit is not perfect. Please find the attached
> torsional profiles. My queries are as follows:
> >
> > 1) Is there any way to identify individual dihedrals contribution by
> looking at entire dihedral fitting profile?
> > 2) FFTK does not consider dihedrals containing hydrogens, then how does
> it generate dihedral parameters for those torsionals?
> >
> >
> > Your help will be highly appreciated.
> >
> >
> >
> >
> > --
> > Thanks and Regards,
> > Mohan Maruthi
> > <test_ml.png>
>
>
>
-- Thanks and Regards, Mohan Maruthi
- Next message: John Stone: "Re: Trouble launching VMD 1.9.2 from the dock in Mac OS X el capitan"
- Previous message: Ky Wildermuth: "issue drawing a PBC box centered on a selection"
- In reply to: Mayne, Christopher G: "Re: FFTK dihedral fitting"
- Next in thread: Mayne, Christopher G: "Re: Re: FFTK dihedral fitting"
- Reply: Mayne, Christopher G: "Re: Re: FFTK dihedral fitting"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ] [ attachment ]


