What is VMD?
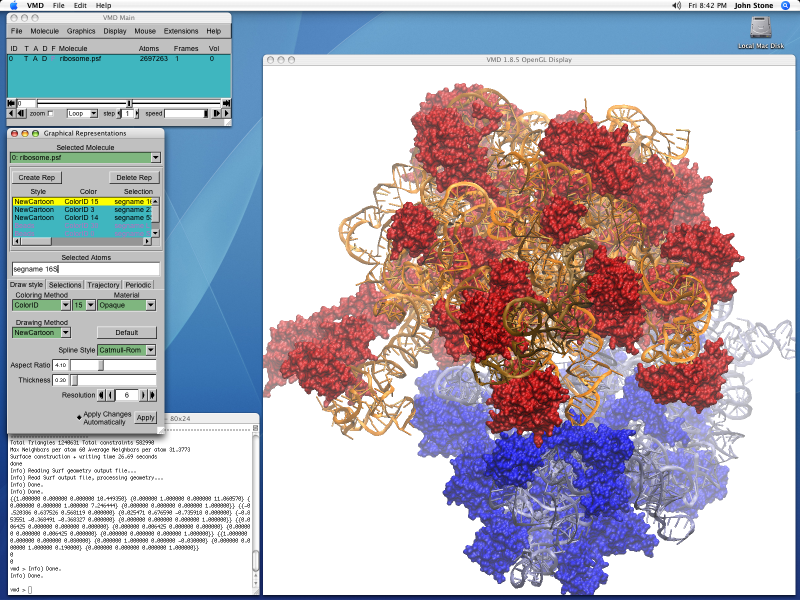
VMD 1.8.5 Screen Shot
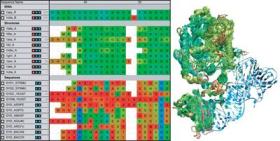
MultiSeq 2.0 Plugin
- Support for all major computer platforms
- Support for multicore processors
- Support for GPU accelerated computation
- Many excellent VMD tutorials developed locally, and by the research community at large
- No limits on the number of molecules, atoms, residues or number of trajectory frames, except available memory
- Many molecular rendering and coloring methods
- Stereo display capability
- Extensive atom selection syntax for choosing subsets of atoms for display (includes boolean operators, regular expressions, and more)
- Support for over 60 molecular file formats and data types through an extensive library of built-in file reader/writer plugins and translators
- VMD includes a multiple sequence alignment plugin, a unified bioinformatics analysis environment that allows one to organize, display, and analyze both sequence and structure data for proteins and nucleic acids.
- Ability to export displayed graphics to files which may be processed by a number of popular ray tracing and image rendering packages, including POV-Ray, Rayshade, Raster3D, and Tachyon.
- User-extensible graphical and text-based user interfaces, built-on standard Tcl/Tk and Python scripting languages
- Extensions to the Tcl language which enable researchers to write their own routines for molecular analysis
- Modular, extensible source code using an object-oriented design in C++, with a programmers guide describing the program architecture and source code
- Integration with the program NAMD, a fast, parallel, and scalable
molecular dynamics program developed in conjunction with VMD
in the Theoretical and Computational Biophysics Group at the
University of Illinois.
See the NAMD WWW home page for more info:
http://www.ks.uiuc.edu/Research/namd/ - VMD works well with projected display systems like the 3-D Projection Facility maintained by the Theoretical and Computational Biophysics Group.
- VMD can be used to concurrently display and interact with a running NAMD simulation.
- Using VMD as a Web/MIME Helper Application
- VMD script library
- Portable and Intuitive GUI
- Interfacing with MD Programs (i.e. SIGMA, NAMD)
- Interactive MD and VR Device Support
- Making Movies with VMD


