MDFF Plugin, Version 0.4
The MDFF plugin provides commands for setting up and analyzing molecular dynamics flexible fitting simulations, i.e., simulations in which an atomic structure is flexibly fitted into a density map. Traditional MDFF simulations fit structures into a cryo-EM density map, while the newer xMDFF method can be used for the refinement of low-resolution x-ray crystallography structures.
Getting started
For a description of the MDFF method, required software, related publications, related updates, etc., please see the MDFF website. The following reference provides a practical guide to MDFF:
Molecular Dynamics Flexible Fitting: A practical guide to combine cryo-electron microscopy and X-ray crystallography.
Leonardo G. Trabuco*, Elizabeth Villa*, Eduard Schreiner, Christopher B. Harrison, and Klaus Schulten. Methods, 49, 174-180, 2009.
MDFF tutorial: For tutorials related to the MDFF method, click here.
Getting help: Any questions, bug reports, feature requests, suggestions or comments related to MDFF should be sent to the VMD mailing list.
Acknowledging MDFF in publications
Please cite the following reference in any work that applied MDFF:
Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics.
Leonardo G. Trabuco*, Elizabeth Villa*, Kakoli
Mitra, Joachim Frank, and Klaus Schulten.
Structure, 16, 673-683, 2008.
Please cite the following reference in any work that applied xMDFF:
xMDFF: Molecular dynamics flexible fitting of low-resolution X-Ray structures.
Ryan McGreevy*, Abhishek Singharoy*, Qufei Li, Jingfen Zhang, Dong Xu, Eduardo Perozo, and Klaus Schulten. Acta Crystallographica, D70, 2344-2355, 2014.
* Equal contribution.
MDFF developers
The MDFF plugin was written by Leonardo Trabuco, Elizabeth Villa, and Ryan McGreevy.
MDFF commands
The current version of the MDFF plugin provides both text commands and a graphical user interface. You first need to load the MDFF plugin by typing package require mdff in the VMD text console or the Tk Console. Typing mdff will print the available subcommands:
Usage: mdff <command> [args...]
Commands:
ccc -- calculates the cross-correlation coefficient
check -- monitors the fitting via RMSD and CCC
constrain -- creates a pdb file for restraining atoms
delete -- deletes volume corresponding to atomic structure
edges -- creates a map with smooth edges
fix -- creates a pdb file for fixing atoms
griddx -- creates a map for docking
gridpdb -- creates a pdb file with atomic masses in the beta field
setup -- writes a NAMD configuration file for MDFF
sim -- creates a simulated map from an atomic structure
Typing each command will print the corresponding usage information (e.g., type mdff sim to get the usage information of the sim subcommand.
mdff ccc
Usage: mdff ccc <atom selection> -i <input map> -res <map resolution in Angstroms> ?options?
Options:
-spacing <grid spacing in Angstroms> (default: 1.0)
-threshold <x sigmas>
-allframes (average over all frames)
mdff check
Usage: mdff check ?options?
Options:
-mol <molid> (default: top)
-frames <begin:end> or <begin:step:end> or all or now (default: all)
-rmsd -- calculate the RMSD with respect to the refence structure
-rmsdseltext <selection text for calculating the RMSD> (default: backbone)
-refpdb <pdb file> -- reference for RMSD calculation (default: frame 0)
-rmsdfile <file to write RMSD> (default: none)
-ccc -- calculate cross correlation coefficient
-map <input map> (required by -ccc)
-res <map resolution in Angstroms> (required by -ccc)
-spacing <grid spacing in Angstroms> (default: 1.0)
-cccseltext <selection text for calculating the cross correlation (default: protein or nucleic)
-cccfile <file to write cross correlation> (default: none)
-threshold <x sigmas>
mdff constrain
Usage: mdff constrain <atomselection> -o <pdb file> ?options?
Options:
-col <column> (default: B)
-k <force constant in kcal/mol/A^2> (default: 10.0)
mdff delete
Usage: mdff delete <atom selection> -i <input map> -o <output dx map> ?options?
Options:
-res <target resolution in Angstroms> (default: 10.0)
-spacing <grid spacing for "mask" map in Angstroms> (default: 1.0)
-allframes -- average over all frames
mdff edges
Usage: mdff edge -i <input map> -o <output dx map> ?options?
Options:
-kernel <Gaussian kernel> (default: 3.0)
mdff fix
Usage: mdff fix <atomselection> -o <pdb file> ?options?
Options:
-col <column> (default: O)
mdff griddx
Usage: mdff griddx -i <input map> -o <output dx map> ?options?
Options:
-threshold <value> (default: 0)
mdff gridpdb
Usage: mdff gridpdb -psf <input psf> -pdb <input pdb> -o <output pdb> ?options?
Options:
-seltext -- atom selection text (default: noh and (protein or nucleic))
mdff setup
Usage: mdff setup -o <output prefix> -psf <psf file> -pdb <pdb file> -griddx <griddx file> ?options?
Options:
-gridpdb -- pdb file for docking (default: -pdb)
-diel -- dielectric constant (default: 80; 1 with -pbc)
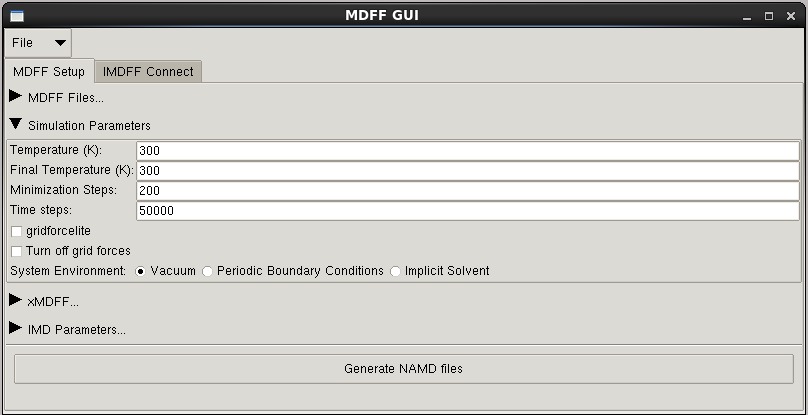
-temp -- temperature in Kelvin (default: 300)
-ftemp -- final temperature (default: 300)
-gscale -- scaling factor for the grid (default: 0.3)
-extrab -- extrabonds file (default: none)
-conspdb -- pdb file with constrained atoms (default: none)
-conscol -- force constant column in conspdb (default: beta)
-fixpdb -- pdb file with fixed atoms (default: none)
-fixcol -- column in fixpdb (default: occupancy)
-scal14 -- 1-4 scaling (default: 1.0)
-step -- docking protocol step (default: 1)
-parfiles -- parameter file list
-minsteps -- number of minimization steps (default 200)
-numsteps -- number of time steps to run (default: 500000)
-margin -- extra length in patch dimension during simulation (default: 0)
-pbc -- use periodic boundary conditions (for explicit solvent)
-gbis -- use generalized Born implicit solvent (not compatible with -pbc)
-dir -- Working Directory (default: current directory)
--lite -- use girdforcelite, a faster but less accuracte calculation of mdff forces
--gridoff -- turn off gridforces, so no mdff forces are applied
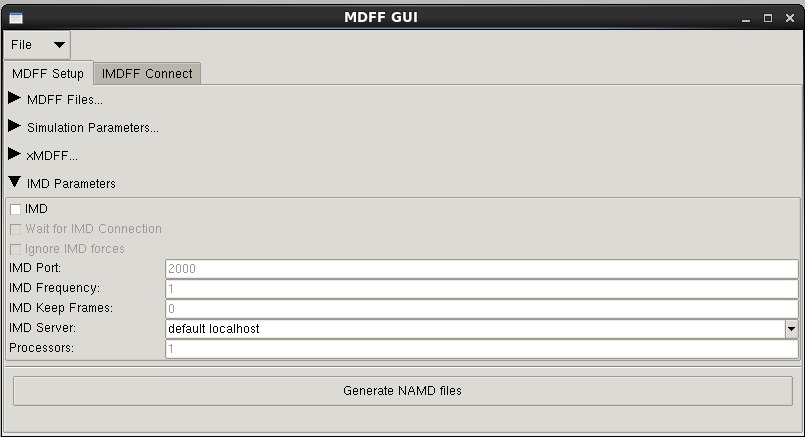
--imd -- turn on Interactive Molecular Dynamics (IMD)
-imdport -- port for IMD connection
-imdfreq -- timesteps between sending IMD coordinates
--imdwait -- wait for IMD connection
--imdignore -- ignore steering forces from VMD
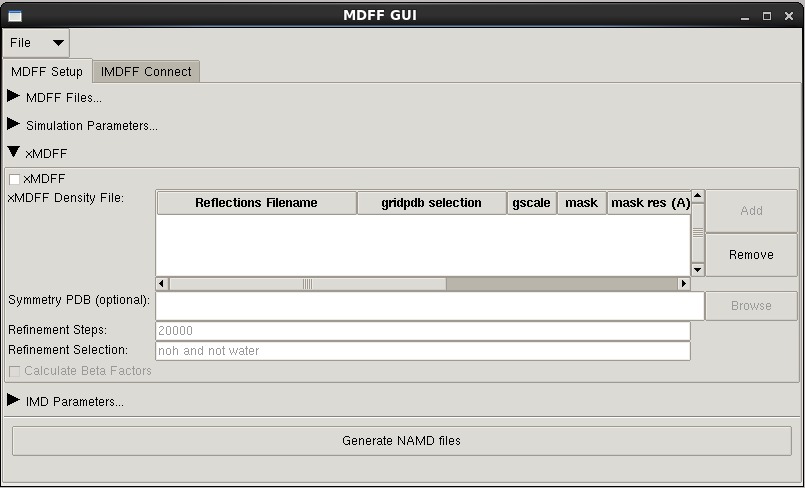
--xmdff -- set up xMDFF simulation. The following options apply to xMDFF only.
-refs -- reflection data file (mtz or cif). Required for xMDFF
-refsteps -- number of refinement steps between map generation (default: 20000)
-crystpdb -- text file (can be PDB) with PDB formatted CRYST line to supply symmetry information (default: none, but recommended)
--mask -- clean generated maps by applying a binary mask around structure (default: off)
-mask_res -- resolution of mask density in Angstroms (default: 5)
-mask_cutoff -- cutoff distance of mask density in Angstroms (default: 5)
--bfs -- calculate beta factors during every map generation step (default: off)
-averagemap -- generate density maps which are averaged from multiple previous steps (default: 0 (off). 1 for on. One entry per map defined in -refs)
-bsharp -- apply beta factor sharpening (or smoothing) to the map(s) (default: 0 (off). Negative values for smoothing, positive for sharpening, 'a' for automatic determination (sharpening only))
mdff sim
Usage: mdff sim <atomselection> -o <output map> ?options?
Options:
-res <target resolution in Angstroms> (default: 10.0)
-spacing <grid spacing in Angstroms> (default: 1.0)
-allframes (average over all frames)
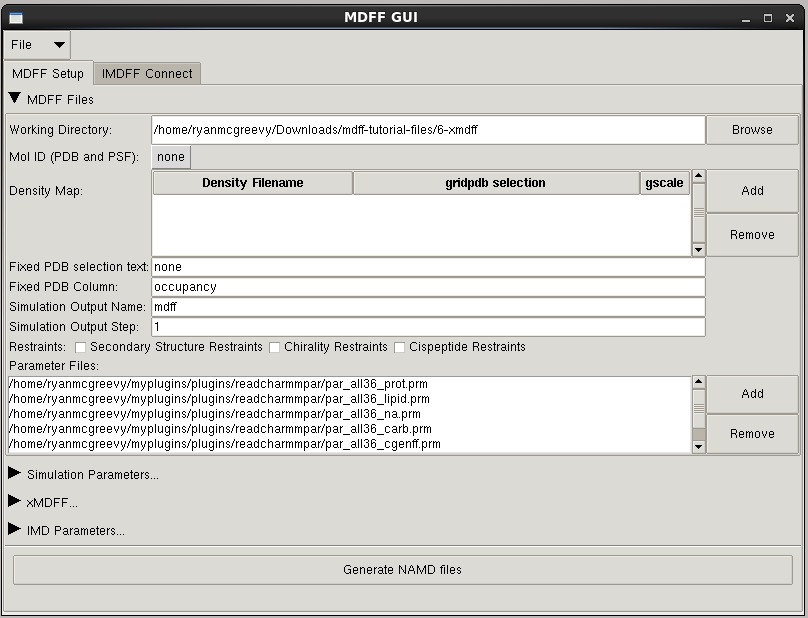
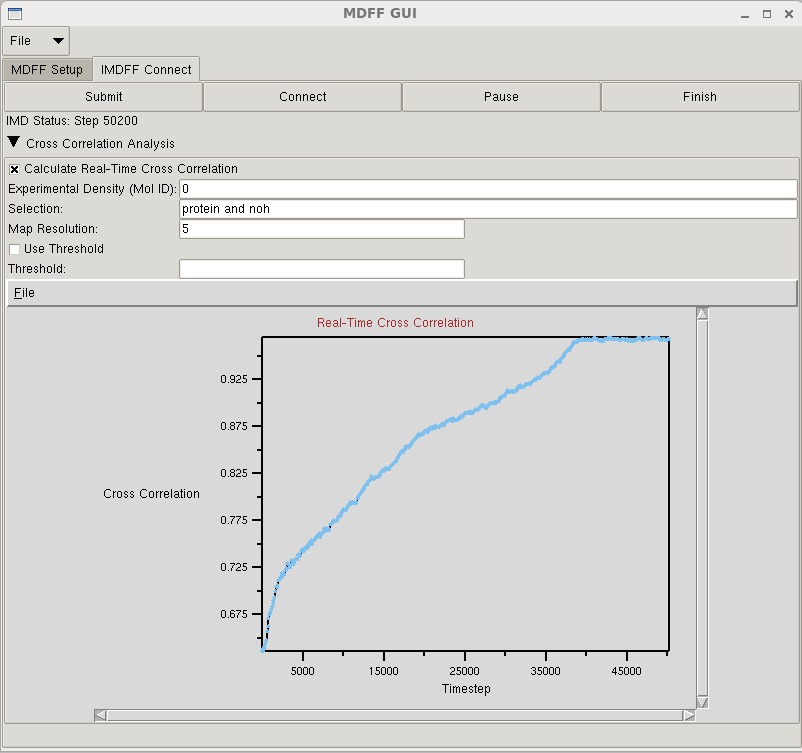
MDFF GUI
The mdff plugin can also be used through a graphical user interface, found in the "Modeling" section of the VMD "Extensions" menu. This interface provides many of the same features found in the command line version of the plugin in a more user friendly package. The GUI can be used to easily set up both MDFF and xMDFF simulations, as well as launch, connect to, and analyze interactive MDFF simulations.