HBonds Plugin, Version 1.2
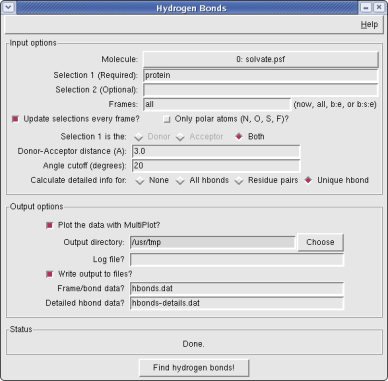
The Hbonds plugin counts the number of hydrogen bonds formed throughout a trajectory. The search can be restricted to a single selection or between two distinct selections, as well as a frame range given by the user.
Criteria for the formation of a hydrogen bond
A hydrogen bond is formed between an atom with a hydrogen bonded to it (the donor, D) and another atom (the acceptor, A) provided that the distance D-A is less than the cut-off distance (default 3.0 Angstroms) and the angle D-H-A is less than the cut-off angle (default 20 degrees).
Options
The plugin provides options for up to two selections (which should not overlap) as well as the frames over which to calculate. The selections can be updated every frame, but at the cost of speed; details about selections that may require this option can be found on the Salt Bridges plugin page. The number of hydrogen bonds vs. time can be plotted immediately within VMD, and/or saved to a file (default hbonds.dat). Additionally, all messages can be output to a log file.
Further options are provided to control the atoms used in the selections (only polar atoms vs. all atoms as normally done in VMD), whether to limit the first selection to be the donor, the acceptor, or both (default: both), and whether to calculate detailed information about the hydrogen bonds. The detailed output includes all hydrogen bonds formed in the trajectory (according to some basic criteria) and their frequency. Note that when using the option "all" for detailed output, the frequency of interaction may be greater than 100%, because a given residue pair may contain more than one hydrogen bond, each of which is counted separately.
Command-line interface
All the functionalities of the plugin are also available through the command line interface.
Usage: hbonds -sel1 <atom selection> <option1> <option2> ...
Options:
-sel2 <atom selection> (default: none)
-writefile <yes|no> (default: no)
-upsel <yes|no> (update atom selections every frame? default: yes)
-frames <begin:end> or <begin:step:end> or all or now (default: all)
-dist <cutoff distance between donor and acceptor> (default: 3.0)
-ang <angle cutoff> (default: 20)
-plot <yes|no> (plot with MultiPlot, default: yes)
-outdir <output directory> (default: current)
-log <log filename> (default: none)
-writefile <yes|no> (default: no)
-outfile <dat filename> (default: hbonds.dat)
-polar <yes|no> (consider only polar atoms (N, O, S, F)? default: no)
-DA <D|A|both> (sel1 is the donor (D), acceptor (A), or donor and acceptor (both))
Only valid when used with two selections, default: both)
-type: (default: none)
none--no detailed bonding information will be calculated
all--hbonds in the same residue pair type are all counted
pair--hbonds in the same residue pair type are counted once
unique--hbonds are counted according to the donor-acceptor atom pair type
-detailout <details output file> (default: stdout)
Authors
JC Gumbart
and Dong Luo (us917_at_yahoo.com)


