All cells share a universal, minimal set of biochemical processes, essential to all life. The search for defining this minimal set has lead to the synthetic assembly of the first minimal cell, JCVI-syn3.0: a cell with only 473 genes, the bare minimum requirements for independent cellular life. Its genetic basis was Mycoplasma mycoides; the Mycoplasmas have long been of interest to investigate fundamentals of life due to their evolutionarily reduced genomes, and at the same time represent an important class of pathogens, implicated in diseases such as pneumonia, urogenital diseases and certain types of cancer.
Thus, JCVI-syn3.0 constitutes a platform to study the function of every gene that is essential for cellular life \cite{HUTC2016in silico model of the entire minimal cell at all-atom resolution within reach.
This model will enable a spatially resolved study of the biochemical processes fundamental to life.
At the same time, it will allow the computational analysis of cell-scale effects resulting from biomedically relevant
perturbations (e.g., antibiotics) on this pathogen prototype.
Here, using the proposed technological advances of the Center, as well as planned exascale machines, we will study:
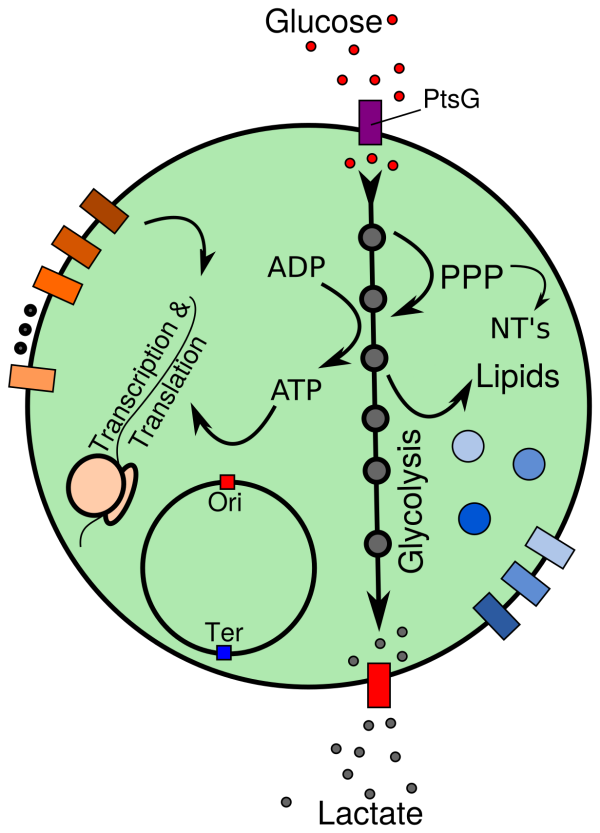
(1) the complete, minimal network of cellular processes, i.e. DNA replication, transcription, translation, ribosome assembly and metabolism,
(2) the physical properties of the crowded cytoplasm and cell membrane within the entire JCVI-syn3.0 cell, and their coupling to the network of cellular processes
and (3)
biomedical applications, including: molecular and cell-scale effects of antibiotics; the evolution of new metabolic functions, to reconstruct the emergence of virulence; and the potential implementation of new biosynthetic pathways to produce biomedical compounds of interest.
Innovation
There has never been an atomistic representation of an entire living cell before; nor has there been a complete, spatially resolved, in silico representation of all reaction networks that make up a cell. The former is needed to probe motions of and interactions between macromolecular components, the latter to model how behavior of individual cytoplasmic and membrane components gives rise to cellular life as a whole.
The first major challenge towards a detailed in silico evaluation of JCVI-syn3.0 arises from the all-atom modeling of its membrane and cytoplasm.
JCVI-syn3.0 spans ~0.4 micrometer in diameter, and contains
~500,000 base pairs of DNA, RNA totaling ~3,000,000 bases, ~150 ribosomes, ~300,000 protein molecules and ~3 million lipid molecules, adding up to ~4 billion atoms.
To address the modeling challenge, we need to obtain structures of several hundred different transmembrane and cytoplasmic proteins;obtaining all these structures by conventional homology modeling is immensely tedious and time-consuming and drives the development of ModelMaker (TRD3).
Furthermore, construction of a realistic membrane model is currently not feasible at the micrometer scale, requiring scalable protocols for lipid assembly and protein embedding; these protocols will be provided with the Cell-Membrane (TRD3) and Lipid Shrink Wrap (TRD3) tools in the Cellular Membrane Modeling (CMM) suite.
Placement of membrane and cytoplasmic proteins together with DNA and RNA within the cell is difficult (see studies on modeling a portion of bacterial cytoplasm and will be enabled in an automated fashion with tools for cell-scale placement of macromolecules (TRD2) that also leverage experimental data on cell architecture. The second challenge lies in the necessity of performing nano- to microsecond long MD simulations of the ~4 billion atom cell to probe macromolecular conformations in crowded cellular environments, which is impossible even with petascale computing, and will be enabled by petascale exascale computing (TRD1) techniques developed in NAMD. The third challenge stems from probing the physical properties inside the cell. The required millisecond macromolecular dynamics will be accessed with Atomic Resolution Brownian Dynamics (ARBD) of the cellular system. Setup of a cell-scale BD simulation requires assembly of whole-cell potentials from MD-averaged electrostatic maps of individual cytoplasmic and membrane components, which will be enabled with BDwiz (TRD3). Sampling of millisecond, cell scale macromolecular dynamics requires parallelization of ARBD over multiple GPUs and nodes, to be provided with GPU-BD (TRD3).
This dynamics will also be used to refine the description of macromolecular association and diffusive properties in a reaction-diffusion model of the cellular reaction network, driving the extraction of diffusion parameters and association rates from BD trajectories with PMForge (TRD3). The fourth challenge is the construction and simulation of a Reaction-Diffusion Master Equation (RDME) model in order to probe
the complete, minimal network of cellular processes in the native JCVI-syn3.0 cell. Defining the RDME requires explicit description of thousands of reactions, involving 100,000s of enzymes, represented by simulation particles totalling millimolar concentrations. Because earlier versions of LM were aggressively optimized for smaller cellular networks, this minimal whole-cell simulation will necessitate increasing limits on the numbers of species and reactions and allowing for higher particle densities with BIG-LM (TRD3) and fully exploiting MPI parallelization with MLP-LM (TRD3). An additional obstacle arises in the RDME description of metabolites that exist in high concentration (e.g., mM of ATP) across the minimal cell;
at such high concentrations the usual particle representation in RDME becomes unnecessary. The high-concentration metabolites will be handled via a continuous representation connected to the RDME in Multiscale-LM (TRD3). The fifth challenge is posed by visualization and analysis of all three representations of the whole JCVI-syn3.0 cell. The necessity to interactively navigate and inspect a billion-atom system drives large system visualization (TRD2) functionalities in VMD.
In addition, such a visualization could not be accomplished on a workstation. Remote visualization on supercomputers will hence be enabled in VMD (TRD2). Finally, the RDME model of all cellular processes requires visualization semantics for components like particles in changing quantity and spatial regions. These requirements drive enhancements for cellular visualization and analysis (TRD2) in VMD.
Approach
The Center boasts a broad range of expertise on modeling biological systems from atomistic simulations of large macromolecular assemblies, to coarse-grained models of biomolecular structures and processes, to reaction-diffusion descriptions of cellular behavior up to system-level analyses of cell populations. This expertise will be leveraged to create a detailed model for a whole JCVI-syn3.0 cell using multi-scale dynamics: microseconds with MD, milliseconds with BD and hours with RDME. The feedback between these methods will both serve to iteratively refine each individual representation as well as allow for a multi-level characterization of the whole cell. In particular, the model will reveal the physical properties of the cytoplasm and its coupling with
the minimal-cell reaction network both in its natural state and under the influence of perturbations (e.g., drugs) or modifications (e.g., new pathways/functions). As a first step towards an RDME description of JCVI-syn3.0, its genome has been used to reconstruct its metabolic network and derive a steady-state flux-balance analysis (FBA) draft model of JCVI-syn3.0 metabolism. Inclusion of functional characterization of genes not yet annotated (Hutchison), as well as proteomics data (Hutchison), will allow for this model to be refined with enzyme-count based reaction flux constraints, and will allow the study of metabolic variability in a population of JCVI-syn3.0 cells with the Population FBA method developed by the Center. From these detailed FBA studies, a simplified kinetic model of metabolism will be constructed. Combining this metabolic component with an RDME model of transcription and translation developed at the Center , and incorporating cryo-electron tomography data (Villa) to realistically construct the cell geometry, leads to the assembly and simulation of the first genome-scale RDME model, leveraging recent advances in the LM code as well as ongoing expansions (TRD3). This RDME model will be able to probe the complete network of cellular processes in JCVI-syn3.0 with spatiotemporal resolution. It will thus allow both the study of systemic effects of antibiotic compounds (e.g., the emergence of synergistic effects and the interaction of new metabolic pathways with the rest of the network, which would allow one to study virulence and drug production. The underlying molecular details, like the interactions between the proteins forming the networks and the target finding and binding of drugs, will need to be validated via atomistic simulations. An atomic scale model of JCVI-syn3.0 will be described using genomics,
proteomics (Hutchison), and cryo-ET data (Villa), and constructed using the various
advances in VMD (TRD2), MDFF (TRD3) and CMM (TRD3). NAMD (TRD1) simulations of
this model will on the one hand reveal how membrane proteins affect the curvature of the
membrane. On the other hand they will capture the flexibility and non-native-like, yet functional, conformations of the cytoplasmic proteins under the influence of a protein-packed membrane and crowded cytoplasmic environment. Such packing and crowding effects on protein structure can only be captured within a whole-cell model. Noting, however, that protein diffusion, protein-protein and protein-drug association rates are beyond the limits of classical MD, millisecond-scale ARBD (TRD3) simulations will be performed for the cytoplasmic components confined within a fixed membrane. To this end, time-averaged electrostatic maps derived from atomistic simulations will be employed to drive the ARBD simulations. In addition to protein diffusion and association rates, the MD and ARBD simulations can then also provide
an accurate representation of drugs finding their targets, which feeds back into the RDME model.