DBP7: Chromatin
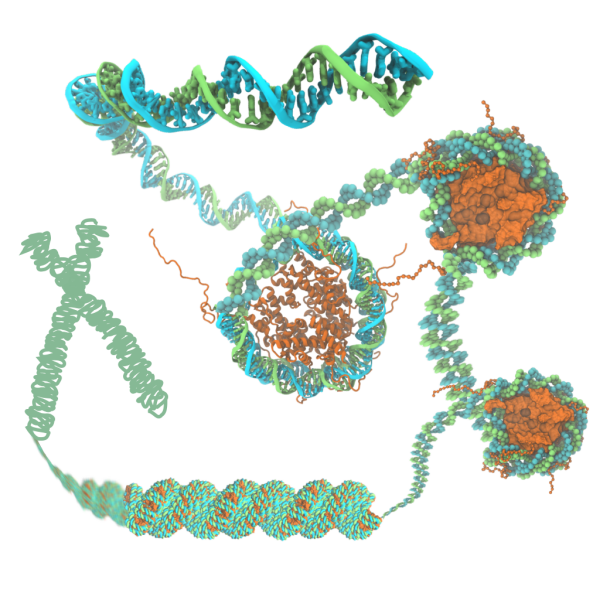
The structure of chromatin plays a pivotal role in gene expression in eukaryotic cells, but nevertheless remains poorly characterized. The basic unit of chromatin is a nucleosome particle formed by wrapping double-stranded DNA around histone proteins. Nucleosomes interact with each other in part through disordered tails. Histone tails are targets for epigenetic modifications that alter chromatin compaction and, correspondingly, gene expression. The structure of chromatin is of particular interest since chromatin remodeling genes, which affect chromatin structure, are implicated in a number of diseases (e.g. Rett syndrome, Rubinstein--Taybi syndrome, alpha thalassemia)~ and in the genesis of many types of cancer. Innovations in computer simulation techniques that combine atomic and coarse-grained (CG) methods will be used to (1) elucidate how physical forces drive chromatin organization and (2) determine the effects of epigenetic modifications on chromatin structure.
The main challenge in simulating chromatin structure is the huge range of length scales spanned by chromatin structure (from ~100~basepair-wrapped histones to kilobase-sized genes to megabase-sized topologically associated domains). Accordingly, small pieces of chromatin have been studied using CG models of the nucleosome. However, each of these models either sacrifices details important for nucleosome interactions or includes too many internal degrees of freedom to allow access to long timescales. To study chromatin structure, we will employ the Center's unique approach to CG, which preserves structural detail while allowing access to long-timescale simulations of large systems. In this approach, the CG parameters are optimized to reproduce data (e.g.~probability densities) from all-atom simulation---a challenge because the conformations of histone tails must be exhaustively sampled over many microseconds in all-atom simulations. Replica exchange MD simulations leveraging single-node enhanced performance (TRD1) will enable efficient sampling of histone tail conformations, allowing the construction of a CG model using the ARBDpmf (TRD3) plugin. The next challenge will be setting up and performing CG simulations of hundreds of thousands of physical particles (representing histone cores) and bead-based polymers (representing histone tails and DNA). The VMD plugin \ARBDwiz\ (TRD3) will assist in the preparation of the simulation systems, while ARBDgpu (TRD3) will run these massive CG simulations efficiently on GPUs. A final challenge is representing the action proteins that cleave DNA to relax and untangle chromatin. ARBDchem (TRD3) will allow us to model such processes.
Our study of chromatin structure will build upon our refined all-atom description of DNA--ion interactions, our expertise on DNA--DNA and DNA--protein interactions and our experience constructing structurally-detailed CG models. The ARBDpmf (TRD3) plugin will be used to distill inter-nucleosome forces suitable for CG simulations from all-atom replica exchange MD simulations that take advantage of single-node enhanced performance (TRD1) in NAMD. CG simulations set up via the \ARBDwiz\ (TRD3) plugin will run on multi-node, GPU-accelerated ARBDgpu (TRD3) and will provide a description of chromatin structure in unprecedented detail. The actions of topoisomerase proteins on chromatin will be modeled through reactions using the ARBDchem (TRD3) feature of the BD code. The sequence-specific flexibility of double-stranded DNA will be accounted for by tuning the CG model against all-atom simulations of a large number (~100) of DNA minicircles with validation from high-throughput sequencing of a looped DNA assay (Ha). Inter-nucleosome interactions in all-atom and small-scale CG simulations will be validated through comparison with single-molecule FRET measurements (Ha). From all-atom simulations, we hypothesize that DNA sequence, DNA methylation and histone modifications, directly modulate inter-nucleosome forces that have implications for chromatin structure on the megabase scale. Observations on the megabase scale will be validated through a mixture of Hi-C data and through bioinformatics analyses (Song).