


Next: High-resolution real-space refinement
Up: Rosetta/MDFF Tutorial
Previous: Model fit to mid-resolution
Contents
Subsections
Homology model
For the 26S proteasome in human, a 3.9 Åcryo-EM density is available, which we will use to reap
the full benefits of our model building tool for high resolutions.
- 1
- Download and install MODELLER
Go to the MODELLER website (https://salilab.org/modeller/tutorial/), download the newest version of MODELLER and request a license. Install MODELLER on your workstation.
- 2
- Sequence alignment
Go to http://www.uniprot.org/align/nd align the sequence of yeast Rpn11 (Uniprot ID P43588) and human Rpn11 (Uniprot ID O00487).
The alignment reveals that the structurally resolved part of the sequence from amino acid 23 to 306 of yeast Rpn11 aligns with the human sequence range of 27-310 with a similarity of 81 %. Download the human Rpn11 fasta sequence and extract the range from amino acid 27 to 310 to a new text file called rpn11_human_27-310.fasta. You can use the aforementioned subrange.py script to do so. For MODELLER input, create the file rpn11_human_27-310.seq by adding the header lines
>P1;rpn11_human_27-310
sequence:::::::::
to the fasta file. Furthermore, append an asterisk (*) to the last amino acid code.
- 3
- Preparing MODELLER run
Create a new folder homology and another folder modeller_run in it. Navigate to the modeller_run directory and copy the
Python scripts from the corresponding tutorial files to it. Furthermore, copy the refined model of yeast Rpn11 rpn11_yeast_midres.pdb,
rpn11_human_27-310.seq,
rpn11_human_27-310.fasta and rpn11_yeast_23-306.fasta to the MODELLER
folder. Make the following changes to the align2d.py script:
- line 5: Change the argument to file='' to rpn11_yeast_midres.pdb.
Execute the align2d.py script by running the following command in the terminal:
mod9.17 align2d.py
- 4
- Running MODELLER
If the alignment script ran without errors, you can build the homology model by simply running
mod9.17 model-single.py
in the terminal to generate a set of 10 homology models. The assess_methods section in model-single.py defines the scoring
functions for the homology model. After the run, we will continue with the model that has the lowest score in the DOPEHR column, specified
in the log file model-single.log.
To position the generated homology model in the Rpn11 human high-resolution density,
we need to perform a rigid body docking. As docking software, we will use the Situs colores tool.
- 1
- Installing the Situs package
Go to the Situs website (http://situs.biomachina.org) and follow the
download and installation instructions. Add the binary folder to your $PATH environment variable.
- 2
- Docking the Rpn11 yeast model to the human density
Create a folder dock_density in the homology model working directory.
Copy the density file rpn11_human_3_3.9_density.mrc from the corresponding tutorial directory to dock_density,
as well as the best homology model PDB structure of the MODELLER run. Rename the MODELLER output PDB to rpn11_human_27-310_notDocked.pdb.
Run the following Situs command:
colores rpn11_human_3_3.9_density.mrc rpn11_human_27-310_notDocked.pdb -res 3.9 -nprocs 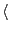 cores
cores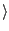
Adjust cores to the number of cores you want to run Situs on.
Rename the output col_best_001.pdb to rpn11_human_27-310.pdb.
In the homology folder, create a folder mdff and navigate to it. Copy the docking output PDB col_best_001.pdb to it, as well as
the density file rpn11_human_3_3.9_density.dx, provided in the tutorial files.
As Situs destroys both correct amino acid numbering and chain identifier, use the script 4.homology/mdff/renumber.tcl to obtain the correctly
labeled PDB file col_best_001-numb.pdb and rename it to rpn11_human_27-310.pdb.
According to section 3.2, interactively fit the
docked structure to the high-resolution density. Save the last frame as rpn11_human_27-310_fit.pdb
To elucidate regions where the structure needs to be refined to the high-resolution density using Rosetta,
you can run a CC coloring as before. You can use the template configuration file in 4.homology/mdff/cccolor to get the following CC-colored files:
- rpn11_human_27-310_fit-ss.pdb: Secondary structure elements are colored by their CC to the density. If a segment does not fit the density, the
CartesianSampler protocol ought to be used to fix this.
- rpn11_human_27-310_fit-res-bb.pdb: The backbone atoms are colored according to their CC with the density. For outliers, we can later on do a backbone refinement to the high-resolution density.
- rpn11_human_27-310_fit-res.pdb: Single residue sidechain outliers can be identified in this pdb file.
With the CC coloring, regions for every single step in the high-resolution density refinement are determined.
Next: High-resolution real-space refinement
Up: Rosetta/MDFF Tutorial
Previous: Model fit to mid-resolution
Contents
www.ks.uiuc.edu/Training/Tutorials