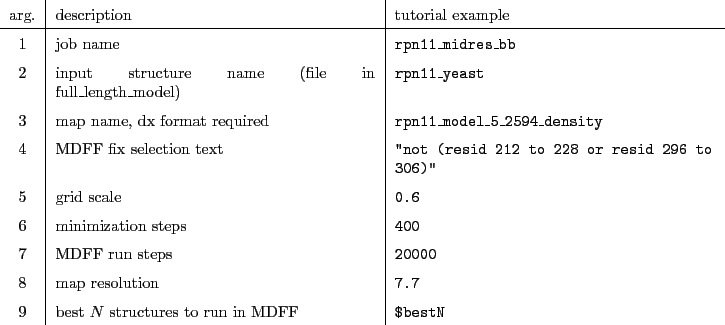
Rigid body docking to the Rpn11 density
Copy the refined Rpn11 yeast model from the last refinement step to the previously created dock_density folder and name it rpn11_yeast_midres.pdb.
As well, copy the rpn11_yeast_3jck_2_6479_density.mrc density file to this folder and navigate to it.
Run the following command in the terminal:
colores rpn11_yeast_3jck_2_6479_density.mrc rpn11_yeast_midres.pdb -res 3.9 -nprocs 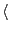 cores
cores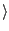
Adjust cores to the number of cores you want to run Situs on.
Rename the output col_best_001.pdb to rpn11_yeast_midres_docked_3jck.pdb.