

Up: Rosetta/MDFF Tutorial
Previous: Homology model
Contents
High-resolution real-space refinement
We will use the ModelMaker plugin with Rosetta and MDFF to refine our homology model fitted to the density to a
final structure, taking advantage of the 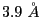 density for Rpn11. This allows a high density weight during the Rosetta runs, as well as high
grid scales for the MDFF runs. In this section, we will perform three different refinement steps, so that we reach a convergence to the density by
increasing a) the Rosetta density weight and b) the grid scale during the corresponding MDFF run.
The refinement steps are:
density for Rpn11. This allows a high density weight during the Rosetta runs, as well as high
grid scales for the MDFF runs. In this section, we will perform three different refinement steps, so that we reach a convergence to the density by
increasing a) the Rosetta density weight and b) the grid scale during the corresponding MDFF run.
The refinement steps are:
- Secondary structure outliers: refinement with CartesianSampler protocol, fitting the given residue ranges to the density + MDFF run
- backbone outliers: the amino acids refined in the first step are further improved by only running a refinement of the backbone atoms + MDFF run
- sidechain refinement of all amino acids with a high Rosetta density weight + MDFF run
As before, we only need one configuration file which we define every refinement step in.
- 1
- Preparing the configuration file
Like in the runs before, create a new folder named highres_refinement and copy the corresponding template configuration file
5.highres_refinement/refine_rpn11_highres.tcl to it.
Create a directory named full_length_model in the highres_refinement folder and
copy the outcoming structure of the last MDFF run to it. Rename the PDB file to rpn11_human_23-310_fit.pdb.
Make the usual changes in the header of the file to match your workstation configuration.
To run NAMD, we need to add a few lines to the file:
- set path <path that contains namd2 executable>
- set topdir <topology and parameter file directory>
- set topfiles <list of topology file names>
- set parfiles <list of parameter file names>
- set ch_seg [list "rpn11" "A" "AP1"]
In the ch_seg variable, we define a list of chains and segnames for PSF generation. The arguments in the list denote [list <name> <chain> <segname>], where the <name> argument is arbitrary. The mutations list can specify a list of mutations for PSF generation. Here, the list elements denote [list <segname> <resid> <new-resname>]. In this case, we don't need any mutations in the PSF, so we define it as an empty list.
The start_rosetta_refine arguments have already been explained in Tab. 6 as well as the MDFF run arguments in Tab. 7.
In this configuration file, we first use the CartesianSampler to refine segments where both backbone and secondary structure need to be refined. You can examine the
residue ranges by CC coloring. The amino acids you want to refine depend on the fitness on your given model to the density. The Rosetta density weight value is increased from step to step, so that the structure more and more converges to its native state, avoiding overfitting in the first place.
start_rosetta_refine_sidechains_density takes the same arguments as the start_rosetta_refine command, except for the CartesianSampler argument
you have to leave out.
- 2
- Running the high-resolution refinement
Run
vmd -dispdev text -e refine_rpn11_highres.tcl
and wait until all steps are finished. The outcome PDB file of every step will be automatically copied to the full_length_model folder.
- 3
- CC coloring of the final model
Use the already explained CC coloring configuration file to compare your input and output models to the last high-resolution refinement step.
Load the colorings of secondary structures, backbone and residues to VMD and compare them. If you further want to refine a range of amino acids, you can iteratively
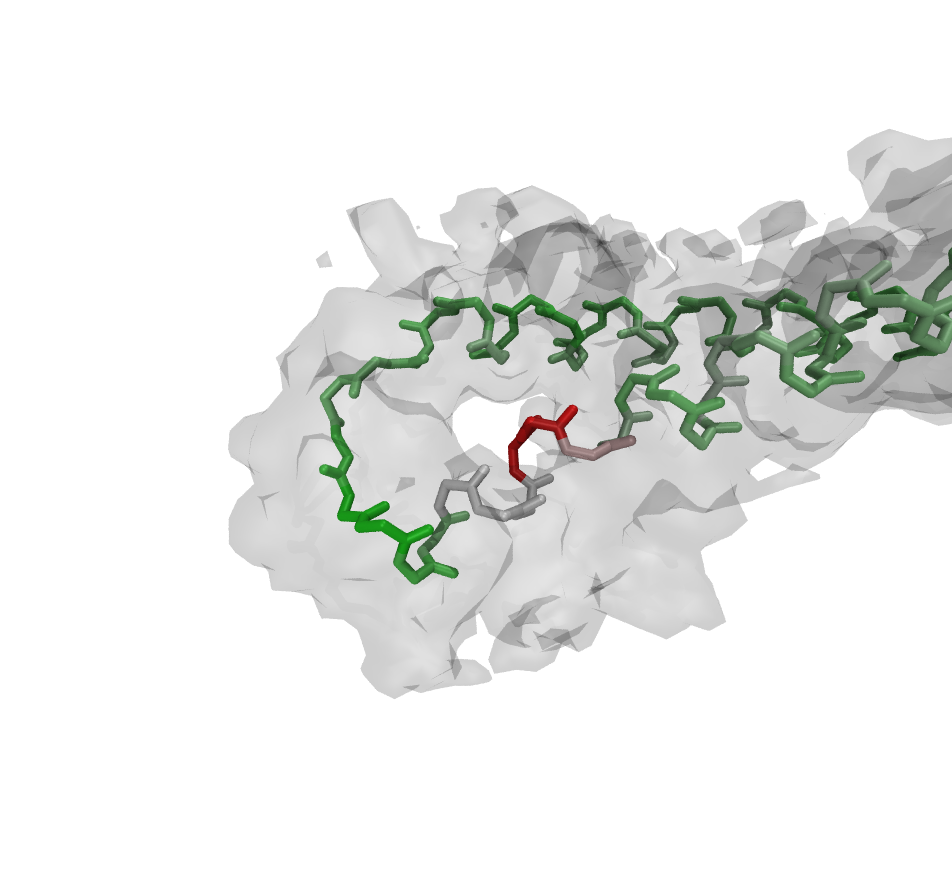
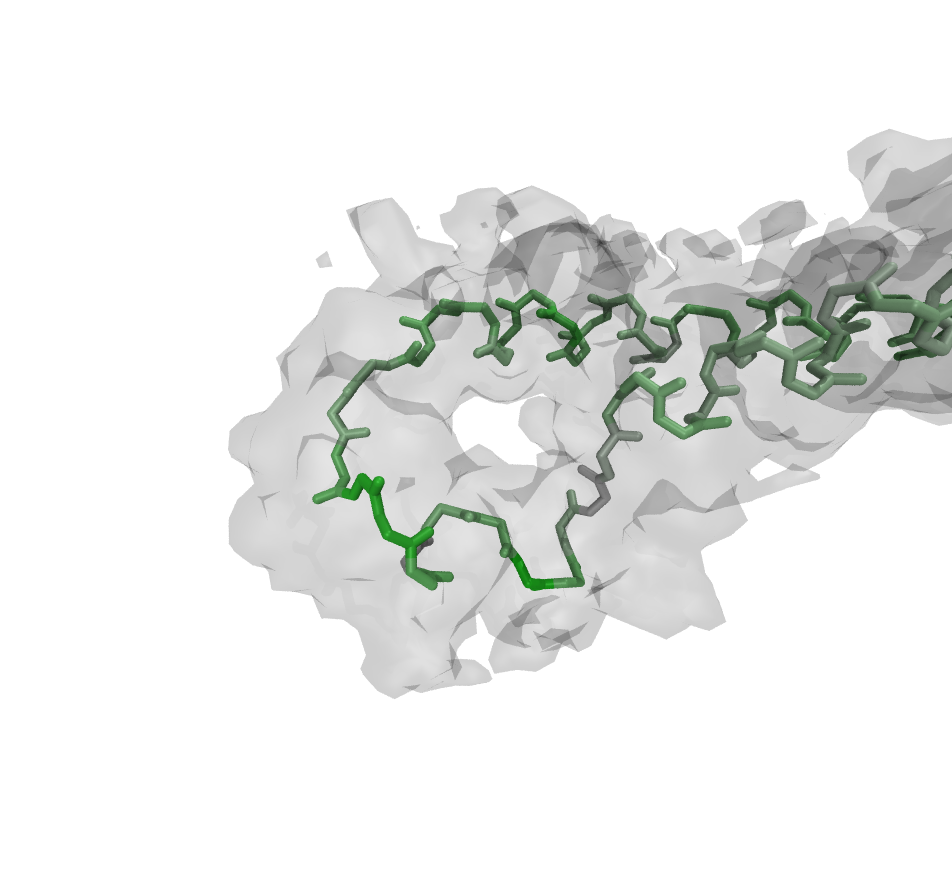
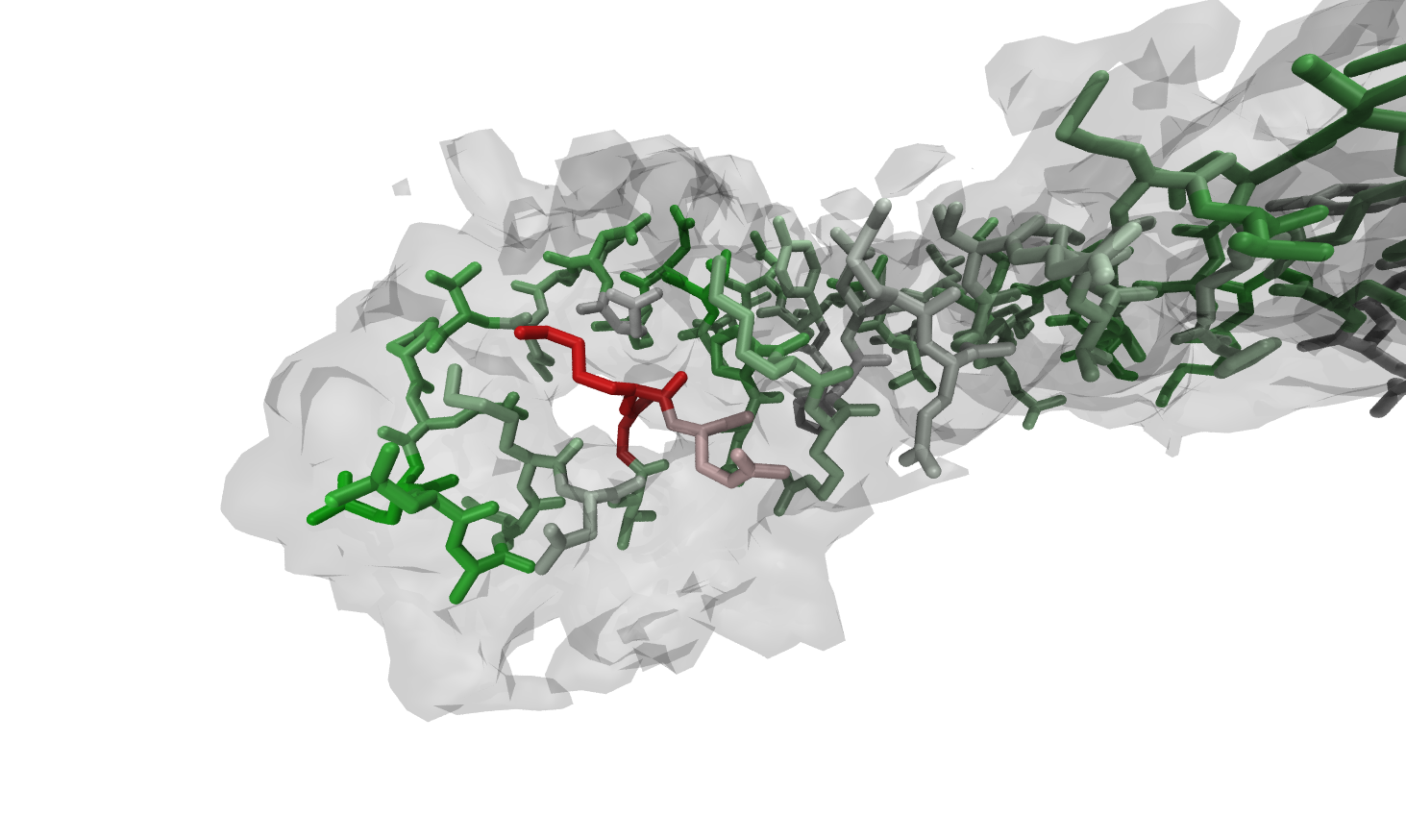
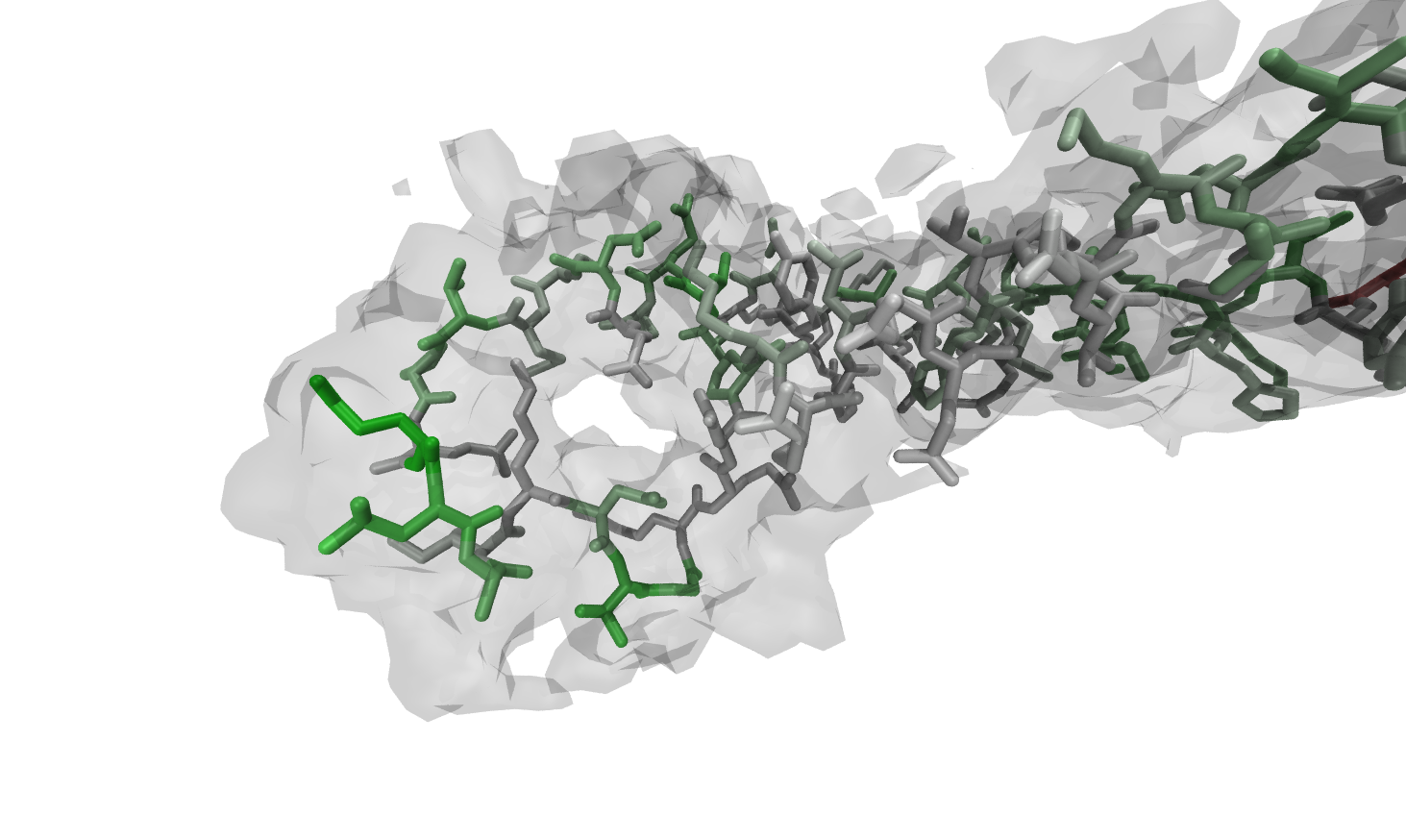
repeat the high-resolution step until you reach the maximum of accuracy. However, Figs. 1 to 4 show the power and usability of the automated refinement with Rosetta and MDFF, wrapped by VMD and simply configurable in one TCL script.
The region around K277 shows low fitness to the high-resolution density before the refinement. After the
protocol run described before, the backbone and the sidechain have adapted a decent conformation due to Rosetta
rotamer optimization, relax steps and MDFF (Figs. 1 to 4).
Figure:
Backbone before refinement
|
|
Figure:
Backbone after refinement
|
|
Figure:
Residues before refinement
|
|
Figure:
Residues after refinement
|
|
Up: Rosetta/MDFF Tutorial
Previous: Homology model
Contents
www.ks.uiuc.edu/Training/Tutorials