NAMD incorporates methods for performing free energy of conformational change perturbation calculations. The system is efficient if only a few coordinates, either of individual atoms or centers of mass of groups of atoms, are needed. The following configuration parameters are used to enable free energy perturbation:
The following sections describe the format of the free energy perturbation script.
These restraints extend the scope of the available restraints beyond that provided by the harmonic position restraints. Each restraint is imposed with a potential energy term, whose form depends on the type of the restraint.
Fixed Restraints
Position restraint (1 atom): force constant ![]() , and reference
position
, and reference
position
![]()
![]()
Stretch restraint (2 atoms): force constant ![]() , and reference
distance
, and reference
distance ![]()
![]()
Bend restraint (3 atoms): force constant ![]() , and reference angle
, and reference angle ![]()
![]()
Torsion restraint (4 atoms): energy barrier ![]() , and reference
angle
, and reference
angle ![]()
![]()
Forcing restraints
Position restraint (1 atom): force constant ![]() , and two reference
positions
, and two reference
positions
![]() and
and
![]()
![]()
![]()
![]()
![]()
Stretch restraint (2 atoms): force constant ![]() , and two reference
distances
, and two reference
distances ![]() and
and ![]()
![]()
![]()
Bend restraint (3 atoms): force constant ![]() , and two reference
angles
, and two reference
angles ![]() and
and ![]()
![]()
![]()
Torsion restraint (4 atoms): energy barrier E![]() , and two reference
angles
, and two reference
angles ![]() and
and ![]()
![]()
![]()
The forcing restraints depend on the coupling parameter, ![]() ,
specified in a conformational forcing calculation. For example, the
restraint distance,
,
specified in a conformational forcing calculation. For example, the
restraint distance, ![]() , depends on
, depends on ![]() , and as
, and as ![]() changes two atoms or centers-of-mass are forced closer together or further
apart. In this case
changes two atoms or centers-of-mass are forced closer together or further
apart. In this case ![]() =
= ![]() , the value supplied at input.
, the value supplied at input.
Alternatively, the value of ![]() may depend upon the coupling parameter
may depend upon the coupling parameter
![]() according to:
according to:
![]() =
=
![]()
Bounds
| Position bound (1 atom): | Force constant |
| and upper or lower reference distance, |
Upper bound:
![]() for
for ![]() , else
, else ![]() .
.
Lower bound:
![]() for
for ![]()
![]()
![]() , else
, else ![]() .
.
![]()
| Distance bound (2 atoms): | Force constant |
| and upper or lower reference distance, |
Upper bound:
![]() for
for
![]() , else
, else ![]() .
.
Lower bound:
![]() for
for
![]() , else
, else ![]() .
.
| Angle bound (3 atoms): | Force constant |
| and upper or lower reference angle, |
Upper bound:
![]() for
for
![]() else
else ![]() .
.
Lower bound:
![]() for
for
![]() else
else
![]()
| Torsion bound (4 atoms): | An upper and lower bound must be provided together. |
| Energy gap |
|
| and angle interval, |
|
|
|
||
|
|
|
||
|
|
|
![]()
Bounds may be used in pairs, to set a lower and upper bound. Torsional bounds always are defined in pairs.
Conformational forcing / Potential of mean force
In conformational forcing calculations, structural parameters such as atomic positions, inter-atomic distances, and dihedral angles are forced to change by application of changing restraint potentials. For example, the distance between two atoms can be restrained by a potential to a mean distance that is varied during the calculation. The free energy change (or potential of mean force, pmf) for the process can be estimated during the simulation.
The potential is made to depend on a coupling parameter, ![]() , whose
value changes during the simulation. In potential of mean force
calculations, the reference value of the restraint potential depends on
, whose
value changes during the simulation. In potential of mean force
calculations, the reference value of the restraint potential depends on ![]() . Alternately, the force constant for the restraint potential may
change in proportion to the coupling parameter. Such a calculation gives the
value of a restraint free energy, i.e., the free energy change of the
syste
. Alternately, the force constant for the restraint potential may
change in proportion to the coupling parameter. Such a calculation gives the
value of a restraint free energy, i.e., the free energy change of the
syste
m due to imposition of the restraint potential.
Methods for computing the free energy
With conformational forcing (or with molecular transformation calculations)
one obtains a free energy difference for a process that is forced on the
system by changing the potential energy function that determines the
dynamics of the system. One always makes the changing potential depend on a
coupling parameter, ![]() . By convention,
. By convention, ![]() can have values
only in the range from
can have values
only in the range from ![]() to
to ![]() , and a value of
, and a value of ![]() corresponds
to one defined state and a value of
corresponds
to one defined state and a value of ![]() corresponds to the other
defined state. Intermediate values of
corresponds to the other
defined state. Intermediate values of ![]() correspond to intermediate
states; in the case of conformational forcing calculations these
intermediate states are physically realizable, but in the case of molecular
transformation calculations they are not.
correspond to intermediate
states; in the case of conformational forcing calculations these
intermediate states are physically realizable, but in the case of molecular
transformation calculations they are not.
The value of ![]() is changed during the simulation. In the first
method provided here, the change in
is changed during the simulation. In the first
method provided here, the change in ![]() is stepwise, while in the
second method it is virtually continuous.
is stepwise, while in the
second method it is virtually continuous.
Multi-configurational thermodynamic integration (MCTI).
In MCTI one accumulates
![]() at several values of
at several values of ![]() , and from these averages
estimates the integral
, and from these averages
estimates the integral
![]()
With this method, the precision of each
![]() can be estimated from the fluctuations of the time
series of
can be estimated from the fluctuations of the time
series of
![]() .
.
Slow growth.
In slow growth, ![]() is incremented by
is incremented by
![]() after each dynamics integration time-step, and the pmf is estimated as
after each dynamics integration time-step, and the pmf is estimated as
![]()
![]()
![]()
Typically, slow growth is done in cycles of: equilibration at ![]() ,
change to
,
change to ![]() , equilibration at
, equilibration at ![]() , change to
, change to ![]() . It is usual to estimate the precision of slow growth simulations from the
results of successive cycles.
. It is usual to estimate the precision of slow growth simulations from the
results of successive cycles.
User-supplied restraint and bounds specifications
urestraint ![]()
n * (restraint or bound specification) // see below
![]()
Restraint Specifications (not coupled to pmf calculations)
| posi | ATOM | kf = KF | ref = (X Y Z) |
| dist | 2 x ATOM | kf = KF | ref = D |
| angle | 3 x ATOM | kf = KF | ref = A |
| dihe | 4 x ATOM | barr = B | ref = A |
Bound Specifications (not coupled to pmf calculations)
| posi bound | ATOM | kf = KF | [low = (X Y Z D) or hi = (X Y Z D)] |
| dist bound | 2 x ATOM | kf = KF | [low = D or hi = D] |
| angle bound | 3 x ATOM | kf = KF | [low = A or hi = A] |
| dihe bound | 4 x ATOM | gap = E | low = A0 hi = A1 delta = A2 |
Forcing Restraint Specifications (coupled to pmf calculations)
| posi pmf | ATOM | kf=KF | low = (X0 Y0 Z0) hi = (X1 Y1 Z1) |
| dist pmf | 2 x ATOM | kf=KF | low = D0 hi = D1 |
| angle pmf | 3 x ATOM | kf=KF | low = A0 hi = A1 |
| dihe pmf | 4 x ATOM | barr=B | low = A0 hi = A1 |
Units
| Input item | Units |
| E, B | kcal/mol |
| X, Y, Z, D | |
| A | degrees |
| kcal/(mol |
The designation ATOM, above, stands for one of the following forms:
A single atom
(segname, resnum, atomname)
Example: (insulin, 10, ca)
All atoms of a single residue
(segname, resnum)
Example: (insulin, 10)
A list of atoms
group ![]() (segname, resnum, atomname), (segname, resnum, atomname),
(segname, resnum, atomname), (segname, resnum, atomname), ![]()
![]()
Example: group ![]() (insulin, 10, ca), (insulin, 10, cb), (insulin,
11, cg)
(insulin, 10, ca), (insulin, 10, cb), (insulin,
11, cg) ![]()
All atoms in a list of residues
group ![]() (segname, resnum), (segname, resnum),
(segname, resnum), (segname, resnum), ![]()
![]()
Example: group ![]() (insulin, 10), (insulin, 12), (insulin, 14)
(insulin, 10), (insulin, 12), (insulin, 14) ![]()
All atoms in a range of residues
group ![]() (segname, resnum) to (segname, resnum)
(segname, resnum) to (segname, resnum) ![]()
Example: group ![]() (insulin, 10) to (insulin, 12)
(insulin, 10) to (insulin, 12) ![]()
One or more atomnames in a list of residues
| group |
| group |
| Examples: | group |
| group |
|
| group |
Note: Within a group, atomname is in effect until a new atomname is used, or the keyword all is used. atomname will not carry over from group to group. This note applies to the paragraph below.
One or more atomnames in a range of residues
| group |
| group |
| Examples: | group |
| group |
|
| group |
The pmf and mcti blocks, below, are used to simultaneously control all
forcing restraints specified in urestraint above. These blocks are performed
consecutively, in the order they appear in the config file. The pmf block is
used to either a) smoothly vary ![]() from 0
from 0 ![]() 1 or 1
1 or 1 ![]() 0, or b) set lambda. The mcti block is used to vary
0, or b) set lambda. The mcti block is used to vary ![]() from 0
from 0 ![]() 1 or 1
1 or 1 ![]() 0 in steps, so that
0 in steps, so that ![]() is
fixed while
is
fixed while ![]() is accumulated.
is accumulated.
Lamba control for slow growth
pmf ![]()
task = [up, down, stop, grow, fade, or nogrow]
time = T [fs, ps, or ns] (default = ps)
lambda = Y (value of ![]() ; only needed for stop and nogrow)
; only needed for stop and nogrow)
lambdat = Z (value of ![]() ; only needed for grow, fade, and
nogrow) (default = 0)
; only needed for grow, fade, and
nogrow) (default = 0)
print = P [fs, ps, or ns] or noprint (default = ps)
![]()
| up, down, stop: | |
| grow, fade, nogrow: | |
| up, grow: | |
| down, fade: | |
| stop, nogrow: | dU/d
|
Lambda control for automated MCTI
mcti ![]()
task = [stepup, stepdown, stepgrow, or stepfade]
equiltime = T1 [fs, ps, or ns] (default = ps)
accumtime = T2 [fs, ps, or ns] (default = ps)
numsteps = N
lambdat = Z (value of ![]() ; only needed for stepgrow, and
stepfade) (default = 0)
; only needed for stepgrow, and
stepfade) (default = 0)
print = P [fs, ps, or ns] or noprint (default = ps)
![]()
| stepup, stepdown: | |
| stepgrow, stepfade: | |
| stepup, stepgrow: | |
| stepdown, stepfade: |
|
For each task, ![]() changes in steps of (1.0/N) from 0
changes in steps of (1.0/N) from 0 ![]() 1
or 1
1
or 1 ![]() 0. At each step, no data is accumulated for the initial
period T1, then dU/d
0. At each step, no data is accumulated for the initial
period T1, then dU/d![]() is accumulated for T2. Therefore, the total
duration of an mcti block is (T1+T2) x N.
is accumulated for T2. Therefore, the total
duration of an mcti block is (T1+T2) x N.
Fixed restraints
| // 1. restrain the position of the ca atom of residue 0. |
| // 2. restrain the distance between the ca's of residues 0 and 10 to 5.2Å |
| // 3. restrain the angle between the ca's of residues 0-10-20
to 90 |
| // 4. restrain the dihedral angle between the ca's of
residues 0-10-20-30 to 180 |
| // 5. restrain the angle between the centers-of-mass of
residues 0-10-20 to 90 |
urestraint ![]()
posi (insulin, 0, ca) kf=20 ref=(10, 11, 11)
dist (insulin, 0, ca) (insulin, 10, ca) kf=20 ref=5.2
angle (insulin, 0, ca) (insulin, 10, ca) (insulin, 20, ca) kf=20 ref=90
dihe (insulin, 0, ca) (insulin, 10, ca) (insulin, 20, ca) (insulin, 30, ca) barr=20 ref=180
angle (insulin, 0) (insulin, 10) (insulin, 20) kf=20 ref=90
![]()
| // 1. | restrain the center of mass of three atoms of residue 0. |
| // 2. | restrain the distance between (the COM of 3 atoms of residue 0) to (the COM of 3 atoms of residue 10). |
| // 3. | restrain the dihedral angle of
(10,11,12)-(15,16,17,18)-(20,22)-(30,31,32,34,35) to 90 |
| // | ( (ca of 10 to 12), (ca, cb, cg of 15 to 18), (all atoms of 20 and 22), (ca of 30, 31, 32, 34, all atoms of 35) ). |
urestraint ![]()
posi group ![]() (insulin, 0, ca), (insulin, 0, cb), (insulin, 0, cg)
(insulin, 0, ca), (insulin, 0, cb), (insulin, 0, cg)![]() kf=20 ref=(10, 11, 11)
kf=20 ref=(10, 11, 11)
| dist | group |
| group |
| dihe | group |
| group |
|
| group |
|
| group |
![]()
Bound specifications
| // 1. | impose an upper bound if an atom's position strays too far from a reference position. |
| // | (add an energy term if the atom is more than 10Å from (2.0, 2.0, 2.0) ). |
| // 2&3. | impose lower and upper bounds on the distance between the ca's of residues 5 and 15. |
| // | (if the separation is less than 5.0Å or greater than 12.0Å add an energy term). |
| // 4. | impose a lower bound on the angle between the centers-of-mass of residues 3-6-9. |
| // | (if the angle goes lower than 90 |
urestraint ![]()
posi bound (insulin, 3, cb) kf=20 hi = (2.0, 2.0, 2.0, 10.0)
dist bound (insulin, 5, ca) (insulin, 15, ca) kf=20 low = 5.0
dist bound (insulin, 5, ca) (insulin, 15, ca) kf=20 hi = 12.0
angle bound (insulin, 3) (insulin, 6) (insulin, 9) kf=20 low=90.0
![]()
| // torsional bounds are defined as pairs. this example specifies upper and lower bounds on the |
| // dihedral angle, |
| // The energy is 0 for: | -90 |
< |
< 120 |
| // The energy is 20 kcal/mol for: | 130 |
< |
< 260 |
| // Energy rises from 0 |
urestraint ![]()
dihe bound (insulin 1) (insulin 2) (insulin 3) (insulin 4) gap=20 low=-90 hi=120 delta=10
![]()
Forcing restraints
| // a forcing restraint will be imposed on the distance between the centers-of-mass of residues (10 to 15) and |
| // residues (30 to 35). low=20.0, hi=10.0, indicates that the
reference distance is 20.0at |
urestraint ![]()
| dist pmf | group |
| group |
![]()
| // 1. during the initial 10 ps, increase the strength of the
forcing restraint to full strength: 0 |
| // 2. next, apply a force to slowly close the distance from
20 to 10 ( |
| // 3. accumulate dU/d |
| // 4. force the distance back to its initial value of 20
( changes from 1 |
pmf ![]()
task = grow
time = 10 ps
print = 1 ps
![]()
pmf ![]()
task = up
time = 100 ps
![]()
pmf ![]()
task = stop
time = 10 ps
![]()
pmf ![]()
task = down
time = 100 ps
![]()
| // 1. | force the distance to close from 20
to 10 in 5 steps. ( |
| // | at each step equilibrate for 10 ps,
then collect dU/d |
| // | ref = 18, 16, 14, 12, 10 , duration = (10 + 10) x 5 = 100 ps. |
| // 2. | reverse the step above ( |
mcti ![]()
task = stepup
equiltime = 10 ps
accumtime = 10 ps
numsteps = 5
print = 1 ps
![]()
mcti ![]()
task = stepdown
![]()
Gradient for position restraint
![]()
![]()
![]()
Gradient for stretch restraint
![]()
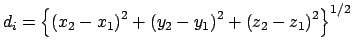
![]()
for atom 2 moving, and atom 1 fixed
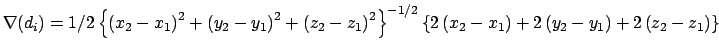
![]()
![]()
Gradient for bend restraint
![]()
Atoms at positions A-B-C
distances: (A to B) = c; (A to C) = b; (B to C) = a;
![]()
![]()
![]()
for atom A moving, atoms B & C fixed (distances b and c change)
![]()
![]()
![]()
for atom B moving, atoms A & C fixed (distances a and c change)
![]()
![]()
![]()
for atom C moving, atoms A & B fixed (distances a and b change)
![]()
![]()
![]()
Gradient for dihedral angle restraint
![]()
Atoms at positions A-B-C-D
![]()
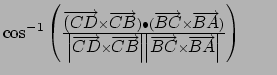 AND
AND
![]()
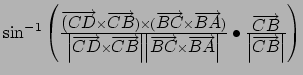
![]()
![]()
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
![]()
|
|
|
|
|
|
|
|
for atom A moving, atoms B, C, & D fixed
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
for atom B moving, atoms A, C, & D fixed
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Gradient for forcing position restraint
![]()
![]()
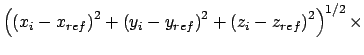 |
||
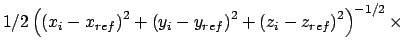 |
||
|
|
![]()
Gradient for forcing stretch restraint
![]()
![]()
![]()
Gradient for forcing bend restraint
![]()
![]()
![]()
Gradient for forcing dihedral restraint
![]()
![]()
![]()