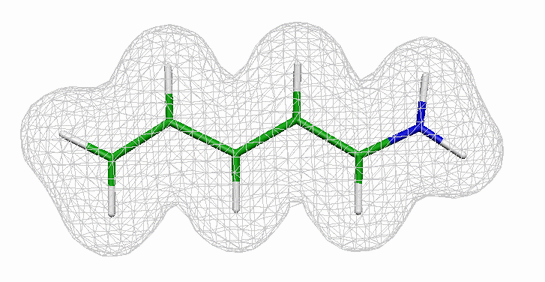
The model system that we will use to introduce you to quantum mechanical (QM) simulations is shown in the figure on the right (the gray wire mesh surface indicates the electron density). The molecule is a retinal analogue and consists of one nitrogen, five carbon and eight hydrogen atoms, which are connected to each other in a conjugated polyene chain. The actual retinal molecule has a longer chain and is covalently bound to a lysine residue of its host protein via a Schiff base linkage. Retinal is the light absorbing moiety in the protein rhodopsin, which is responsible for color vision in the retina of our eyes.
Before you can start studying the properties of the retinal analogue system you need to have a well minimized conformation of your system. You might e.g. be interested in the charge distribution and the equilibrium bond lengths. Since the optimization calculation can take up to 10 minutes you will first start the minimization run and use the meantime to acquaint yourself with the system. All the files you need for part 1 are in the directory QM_tutorial/part1/. Go there now
tbss> cd part1
All paths in the remainder of part 1 will be relative to this directory! To start your minimization cd into the directory min631G.
tbss> cd min631G
Your working directory should contain the file retmin.inp (and sample output files in the sample subdirectory). The Z-matrix in the $DATA field of retmin.inp contains the starting conformation of the retinal analogue which was created "by hand" using standard values for bonds, angles and dihedreals. It does, therefore, not represent the conformation of minimal energy. This is the input file we looked at in the introduction section (have a look with nedit or your favorite editor!). Before you start the minimizations, open a second terminal window and go into your working directory. You can now start the minimization using the command
tbss> rungms retmin.inp >& retmin.log
The simulation will start running and your terminal will become inactive until the computation is finished. The simulation should be finished in less then 15 minutes. Make sure to check, in case the shell is still unresponsive after this amount of time has passed. In your second terminal window you should cd into the same directory where you started the GAMESS run and use the command
tbss> tail -f retmin.log
to view the last 10 lines of the log file (use CNTRL C to exit the tail command). If your job runs correctly you will see a large amount of output which is being generated.
NOTE: If you are running GAMESS via
tbss> rungms filename.inp >& filename.log
in a directory that already contains the files filename.log, filename.dat or filename.F* (e.g. from a previous run) it will terminate. You have to either remove or rename all files besides filename.inp to submit a new simulation in this particular directory. You can remove files with the rm filename command.
Now go back into your main directory by typing
tbss> cd ..
Since the minimization will take a few minutes let us have a look at the retinal analogue molecule. You will not use VMD for this purpose, but rather the quantum chemistry visualization tool MOLDEN to look at GAMESS output.
First, go into the directory QM_tutorial/part1/pdbfile, which contains the pdb file of the structure you are currently minimizing. Type
tbss> cd pdbfile
Then start up MOLDEN by typing
tbss> molden
Note that MOLDEN will "hijack" the terminal in which it was started, until you terminate your MOLDEN session. After typing the command you should see two windows: The main MOLDEN window and the control panel shown below.
|
To load the pdb file of the retinal analogue into MOLDEN press the READ button. This will open a file dialogue box where you can select the file ret.pdb. Please klick on it. The molecule should show up in your main window and you can now close the dialogue box. The nice thing about MOLDEN is that it is also able to read the log file output generated by GAMESS and display geometries, trajectories and other useful information. The buttons in the Draw Mode section allow you to adjust the view to the way you prefer it. Try the Solid, StickColor and Shade buttons to obtain a view you like. Using your mouse in the MOLDEN window you can rotate the molecule. If you press the lower right button in the Zoom menu you are in the "sticky mode" and the molecule rotation will now follow your mouse. Take a few minutes to get acquainted to MOLDEN so you know what you can do with it. Try to avoid pressing the icons in the central column unless you know what you would like to do. |
Once you feel comfortable with MOLDEN have a closer look at your system. After you loaded the pdb file of the retinal analogue into MOLDEN your main window will show something like below (yours may look different depending on your Draw Style and they way you rotated the molecule!)
The retinal analogue consists of 5 carbon, 1 nitrogen and 8 hydrogen atoms. To practice your understanding of internal coordinates you can use MOLDEN’s geometry tools to confirm the Z-matrix input of the $DATA field as discussed in a previous section. Hence measure some of the bonds, angles and dihedrals and compare with the values given in the input file. Note that the starting conformation of the retinal analogue is non-planar.
Now quit MOLDEN by hitting the SKULL button in the lower center of the control panel. Then go back into the main directory by typing
tbss> cd ..
Once your simulation has finished, the shell in which you started the job will become responsive again. Before you do anything else you have to make sure that your simulation terminated normally and everything went smooth (it is a very common mistake to skip this step and take an improperly converged result as final result; always check your log file thoroughly!). First check your log file for successful job completion (you can use any editor of your choice; however, you need to know how to use it to search for strings, since you have to go through the rather lengthy log files. In nedit you can search with the Search -> Find menu item. Xemacs also has a simple interface to search a file in case you don’t know how this feature works in your editor: You can either go to Edit -> Search or press CTRL S and then enter the string you are looking for at the bottom of the window). The last couple of lines should look similar to
EXECUTION OF GAMESS TERMINATED NORMALLY HH:MM:SS DD-MMM-19YY DATA SERVER STATS: TOTAL DISTRIBUTED MEMORY USED (MEMDDI)= 0 MWORDS. FIRST DATA SERVER’S MAXIMUM MEMORY= 0 WORDS, CPU= 0.0 SECONDS. ddikick: all processes have ended gracefully. unset echo ----- accounting info ----- Wed May 21 17:43:37 CDT 2003 Files used on the master node dakar.ks.uiuc.edu were: ESC[00m-rw-r--r-- 1 markus tbres 257429 May 21 17:43 ESC[00m.//rethess.datESC[00m -rw-r--r-- 1 markus tbres 1522 May 21 15:39 ESC[00m.//rethess.F05ESC[00m -rw-r--r-- 1 markus tbres 1456752 May 21 17:43 ESC[00m.//rethess.F10ESC[00m -rw-r--r-- 1 markus tbres 3100768 May 21 17:41 ESC[00m.//rethess.F22ESC[00m -rw-r--r-- 1 markus tbres 1522 May 21 15:39 ESC[00m.//rethess.inpESC[00m -rw-r--r-- 1 markus tbres 29985 May 21 17:43 ESC[00m.//rethess.ircESC[00m -rw-r--r-- 1 markus tbres 562043 May 21 17:43 ESC[00m.//rethess.logESC[00m ESC[m7101.060u 29.011s 2:04:02.62 95.8% 0+0k 0+0io 5617pf+0w
Most important is the statement EXECUTION OF GAMESS TERMINATED NORMALLY telling you that everything went fine (at least from the computational point of view, this does not imply that your simulation actually accomplished what you intended it to do). If your simulations did not terminate with this message you have to figure out what went wrong (contact your lab administrator if you are not sure what to do). Since you performed a geometry optimization you also have to check if GAMESS was able to find the equilibrium geometry or if, for some reason, this was not the case. In the log file look for the line
1 ***** EQUILIBRIUM GEOMETRY LOCATED *****
telling you that the geometry optimization was successful. If your simulation was not able to converge to the equilibrium geometry, try to figure out why. Maybe the maximum number of optimization steps requested via NSTEP in the input file was not sufficiently large. After you convinced yourself that your simulation terminated properly your working directory should contain the files retmin.inp, ret_analogue.log and retmin.dat
We can now use MOLDEN to view the output generated by GAMESS during minimization. Make sure that you are in the directory min631G where you performed your minimization. Start up MOLDEN (click here if you are not sure how) and load the log file. MOLDEN allows you to view the sequence of geometries computed during minimization until the minimum was reached; use the three buttons in the Select Point menu: FIRST brings you to the first frame, NEXT to next one and MOVIE plays the whole trajectory. Look at your output and observe the changes in structure as the system approaches its minimal energy conformation. What is the major change and how could you explain this behavior. Try to identify which bonds in the system are double and which are single bonds. Any pattern?
Now that you have obtained the optimized structure of the retinal analogue you can use GAMESS to determine some of the molecular properties you might be interested in. Let us assume that you would like to perform an MD simulation of rhodopsin, which contains retinal in its binding pocket. As you know by now, in order to do so you need to have the force field parameters of retinal, e.g. partial charges, bond lengths, force constants etc.. These are not provided in the standard CHARMM parameter files and you therefore may have to calculate them yourself. The optimized structure of the retinal analogue already provides the equilibrium values for bond lengths, angles and dihedrals as well as values for charges as you will see shortly, but you are still missing all the force constants. During this session you will only consider how to obtain the force constants for the bonds and the partial charges of the atoms. The other parameters like the angle bending terms can also be calculated, but are more laborious. (You may want to use one of your evening labs to work on them. How would you e.g. go about calculating the stiffness of the angles? You may also want to explore how inclusion of electron correlation will affect the results.)
In order to obtain the force constants
for the bonds you will have to calculate the Hessian matrix of
the retinal analogue in its minimized conformation.
This matrix consists of the second derivatives of the potential
energy with respect to the coordinates. Its eigenvectors are the normal modes
of the molecule. The normal modes can be projected onto the bonds of the
molecule to extract their respective vibration frequencies f and force
constants k, which are related via
![]() ,
where
,
where ![]() is
the reduced mass (i.e.
is
the reduced mass (i.e.
![]() )
of the bonded atoms.
)
of the bonded atoms.
Hence, you will have to set up the run for the calculation of the hessian matrix. Please go into the directory hess631G to set up this run and perform your calculations. To obtain the input file for the calculation you can start from the input file you used for the minimization by copying it to a new file rethess.inp.
tbss> cd ../hess631G tbss> cp ../min631G/retmin.inp ./rethess.inp
Open the new file rethess.inp in a text editor and delete the old internal coordinates in the $DATA section (make sure to only delete the coordinates, not the first two non-coordinate lines in the file!). Next you have to insert the new, optimized internal coordinates. They can be extracted from the log file of your minimization run (remember: this file is located in the directory min631G). Hence load retmin.log into your favorite editor and search for the line
1 ***** EQUILIBRIUM GEOMETRY LOCATED *****
in the last third of the output file. Then scroll down a little more until you see the line
THE CURRENT FULLY SUBSTITUTED Z-MATRIX IS.
The next 13 lines represent your optimized Z-matrix. Now copy and paste the lines into your new input file rethess.inp. Finally you have to tell GAMESS that you would like to calculate the Hessian matrix and that you request the projection of the force constants onto the bonds. The first can accomplished by changing the runtyp to RUNTYP=HESSIAN in the $CONTRL group. The projection can be requested by inserting the keyword fields
$FORCE PURIFY=.TRUE. PRTIFC=.TRUE. DECOMP=.TRUE. $END
$ZMAT IZMAT(1)=1,1,2, 1,2,3, 1,2,4, 1,4,5, 1,5,6, 1,6,7,
1,7,8, 1,4,9, 1,5,10, 1,6,11, 1,7,12, 1,8,13,
1,8,14,
2,1,2,3, 2,3,2,4, 2,3,4,5, 2,4,5,6, 2,5,6,7,
2,6,7,8, 2,3,4,9, 2,4,5,10, 2,5,6,11,
2,6,7,12, 2,7,8,13, 2,7,8,14,
3,1,3,2,4, 3,2,3,4,5, 3,3,4,5,6, 3,4,5,6,7,
3,5,6,7,8, 3,2,3,4,9, 3,9,4,5,10,
3,10,5,6,11, 3,11,6,7,12, 3,12,7,8,13
3,12,7,8,14
$END
at the end of the input file. Don’t forget about inserting a single space in front of the $. Once you are done you may compare your file with the templates in the sample directory. Make sure that you have no additional empty lines, tabs etc., otherwise GAMESS might get confused. Now you are ready to submit your simulation to calculate the Hessian matrix of the optimized retinal analogue. In the directory hess631G type
tbss> rungms rethess.inp >& rethess.log
While the simulation is running you should start extracting the atomic charges from the log file of your minimization run. What you need to know are the partial atomic charges of each of the atoms in the system. Quantum mechanics, however, only provides the electronic wavefunction and has no concept of assigning electrons to particular atoms. There is, therefore, no unique way of assigning partial charges with the help of QM calculations and several schemes exist to achieve this task. We will compare the results of three of them, namely Mulliken, Lowdin and ESP charges. The latter are obtained by fitting charges to the electrostatic potential, whereas the first two follow from projecting the electron density onto the nuclei. To extract the Mulliken and Lowdin charges from the log file of your minimization run, open retmin.log in a text editor. Since these charges are calculated at each step of the minimization run you have to make sure to pick the ones for the final minimization step. Hence start to search from the end of the log file and look for
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
Right underneath this line you will find two columns containing the Mulliken and Lowdin charges. If you are not sure that you found the correct entry you can peek here. Make sure to pick the charges and not the populations. The ESP charges you will find almost at the end of the log file below the line titled
ELECTROSTATIC POTENTIAL
(check here if you are not sure). Now you have three sets of partial atomic charges for your system. You should compare them and notice that they are similar, but far from being identical.
Partical charges for force fields are typically determined using so called restrained ESP charges at a HF/6-31G* level of theory, which are closely related to the ESP charges calculated by us. The reason being the fact that they are much less sensitive to a change in basis than the Mulliken or Lowdin charges. You can convince yourself that this is the case by using one of the evening sessions (don’t do it now) to repeat the charge calculation using the HF/6-31+G level of theory, which includes diffuse functions (consult the manual to find the correct keyword. HINT: look in the BASIS section). You may also compare your computed set of partial charges with CHARMM values which were used in an actual MD simulation (CHARMM charges for retinal analogue).
Once the calculation of your Hessian matrix
has finished have a look at the results.
Make sure, as in the case of the minimization, that your
GAMESS run has executed normally and nothing unexpected has happened.
(note that this time you do not need to check for 1 ***** EQUILIBRIUM
GEOMETRY LOCATED ***** since you did not perform an optimization, but
a calculation of the Hessian matrix!)
Use MOLDEN to look at the output file
rethess.log. Hence start up MOLDEN, load the file and
adjust the view of the molecule to your liking. Then press the Norm Mode
button and a window will pop up that will allow you to view the normal modes of the
retinal analogue that you have just calculated. The modes are ordered according to
increasing frequencies (i.e. energy) and you can view them by simply
clicking on them.
You should try to answer the following questions: Why do the first 6 normal modes
have zero frequency and what are they? Can you discern qualitatively what
distinguishes the low frequency modes (i.e. low energy)
from the high frequency ones (i.e. high energy)?
In order to extract the force constants for the bonds which follow from the hessian matrix you just calculated, have a look at the log file rethess.log. Hence, load the file into an editor and look for the lines
--------------------------------------------------------------------
INTERNAL COORDINATE INTRINSIC FORCE CONSTANTS
(HARTREES/BOHR**2) (MDYN/ANG)
--------------------------------------------------------------------
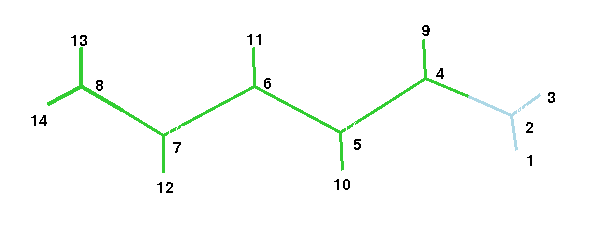
The table right underneath lists the force constants for the 13 bonds in the retinal analogue in the same order as in the Z-matrix in the $DATA section of the input file (note that e.g. STR. 1 2 corresponds to the bond between atom 1 and 2). The figure below gives the atom numbering
.
They are given in
Hartree/Bohr2 and you should convert them into
kcal/(molÅ)2) to be able to use them in a CHARMM
parameter file (note: 1 Hartree = 627.53 kcal/mol,
1 Bohr = 0.529Å).
Your calculation of bond frequencies/force constants
is based on an harmonic approximation for the potential (remember, the
Hessian is the second derivative of the potential energy function!).
This generally overestimates the vibration frequencies and
workers in the field often correct this by multiplying the frequencies f with
by factor of 0.9 (what do you need to multiply the force constants with,
considering that ![]() ?)
?)
Lastly, and quite unfortunately, the CHARMM force constants
are defined via ![]() , therefore
lacking the conventional factor of one half.
Hence, you also have to multiply your force constants by a
factor of 0.5 before you can use them in a CHARMM parameter file.
You can either do the calculations by hand using a calculator, or
copy and paste the 13 lines with the force constants into a text editor
and save them to the file force.dat. You can then use
the command
, therefore
lacking the conventional factor of one half.
Hence, you also have to multiply your force constants by a
factor of 0.5 before you can use them in a CHARMM parameter file.
You can either do the calculations by hand using a calculator, or
copy and paste the 13 lines with the force constants into a text editor
and save them to the file force.dat. You can then use
the command
tbss> gawk '{print $4*0.5*0.81*627.53/(0.529^2)}' force.dat > force_final.dat
to obtain the file force_final.dat with the converted force constants. Use the above figure of the retinal analogue to assign the force constants to the respective bonds and best write them into the figure. Next use MOLDEN to measure the bond distances and also enter them into the figure. Finally, put down the ESP charges of each atom. You have now collected all the information you need to complete the topology and parameter files for the retinal analogue. There is one last thing that you need to consider. In the actual parameter file the bonds between atoms 8-13 and 8-14 have to be the same due to symmetry (the same is true for the bonds between 1-2 and 2-3!). There will hence only be one entry for each type of bond (this is why in the parameter file there are only eleven bond entries to complete and not 13). Since you probably have different values for the force constants and lengths of the bonds 8-13 and 8-14 just take the average! Finally, go back into the main directory
tbss> cd ..
Finally, after you put all that effort into calculating the minimum energy conformation, ESP partial atomic charges, bond lengths, angles etc. you can now put them to use and complete the CHARMM parameter and topology files for our retinal analogue. After generating the psf file using psfgen you can then use NAMD to classically minimize the original conformation of the retinal analogue using your newly designed force field.
Hence go into the directory namdmin/toppar
tbss> cd namdmin/toppar
which contains the parameter and topology files for the retinal analogue (the values you need to fill in are denoted with Xs). Then fill in your calculated ESP charges (they go into the topology file as you know by now; note that the indices of the atoms in the topology file differs slightly from the ones in the Z-matrix and the above figure should help you to identify which correspond to which), the bond lengths and finally the force constants for the bonds that you have calculated (they go into the parameter file! If you have doubts about the correctness of your newly designed files you may peek at at the provided files in the sample directory. If your values differ slightly, that’s all right!) You don’t have to worry about the angle and dihedral terms: leave them as they are. Once you have accomplished this, go into the directory namdmin/psf directory
tbss> cd ../psf
and create the psf file of the retinal analogue (remember: you need the original pdb file and the topology file that you just created; hence copy them into the directory).
tbss> cp ../toppar/top_ret.inp ./ tbss> cp ../../pdbfile/ret.pdb ./
Open VMD and use psfgen to generate the psf file
(you should be able to this yourself by now; in case you run into serious
trouble generating the psf file you may use the create.png
script in the sample directory). Remember: To do so you need the
completed topology file as well as the original pdb file. Please do not close
VMD since we will need it again in a minute.
You are now ready to employ NAMD to minimize the retinal analogue structure
using your own parameter set (1000 minimization steps are more than sufficient).
Please do this in the directory
namdmin/min, hence
tbss> cd ../min/
We encourage you to set up the NAMD input file yourself, but you may also use the provided one in the sample directory. You also need to copy the pdb and psf files from the create_psf directory as well as the completed parameter file from the directory toppar into this directory. Then start the simulation by typing:
tbss> namd2 [name of inputfile] >& [name of outputfile]
The following provides some suggestions for further study during your evening sessions. Please don’t do the exercises during the tutorial, but rather go on with part 2. We encourage you to repeat part 1 using a different, preferably larger, basis set, e.g. HF/6-31G* or HF/6-31+G*. In order to speed up your computations you can start your minimization from the optimized 6-31G geometry. Examine the changes in energy, structure and parameters (charges, bond lengths, force constants) when using a different basis set. This will show you the importance of carefully choosing the basis set of QM calculations in order to obtain meaningful results. It is also important to consider the influence of electron correlation by repeating the above calculations using density functional theory (DFT). Since this will be much more time consuming you will have to do this during one of the evening sessions. In order to employ DFT you simply insert the following line into you input file
$DFT DFTTYP=B3LYP $END
which will use the B3LYP functional to take into account electronic correlation effects.