



Next: Rotations
Up: Defining collective variables
Previous: Contacts
Contents
Index
Subsections
The block rmsd {...} defines the root mean square replacement
(RMSD) of a group of atoms with respect to a reference structure. For
each set of coordinates
 , the colvar component rmsd calculates the
optimal rotation
, the colvar component rmsd calculates the
optimal rotation
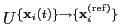 that best superimposes the coordinates
that best superimposes the coordinates
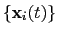 onto a
set of reference coordinates
onto a
set of reference coordinates
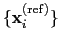 .
Both the current and the reference coordinates are centered on their
centers of geometry,
.
Both the current and the reference coordinates are centered on their
centers of geometry,
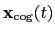 and
and
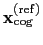 . The root mean square
displacement is then defined as:
. The root mean square
displacement is then defined as:
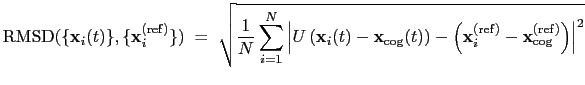 |
(13.5) |
The optimal rotation
is calculated within the formalism developed in
reference [50], which guarantees a continuous
dependence of
with respect to
.
List of keywords (see also ![[*]](crossref.png) for additional options):
for additional options):
-
atoms
 Atom group
Atom group
Context: rmsd
Acceptable values: atoms {...} block
Description: Defines the group of atoms of which the RMSD should be calculated.
Optimal fit options (such as refPositions and
rotateReference) should typically NOT be set within this
block. Exceptions to this rule are the special cases discussed in
the Advanced usage paragraph below.
-
refPositions
Reference coordinates
Context: rmsd
Acceptable values: space-separated list of (x, y, z) triplets
Description: This option (mutually exclusive with refPositionsFile) sets the reference coordinates for RMSD calculation, and uses these to compute the roto-translational fit.
It is functionally equivalent to the option refPositions in the atom group definition, which also supports more advanced fitting options.
-
refPositionsFile
Reference coordinates file
Context: rmsd
Acceptable values: UNIX filename
Description: This option (mutually exclusive with refPositions) sets the reference coordinates for RMSD calculation, and uses these to compute the roto-translational fit.
It is functionally equivalent to the option refPositionsFile in the atom group definition, which also supports more advanced fitting options.
-
refPositionsCol
PDB column containing atom flags
Context: rmsd
Acceptable values: O, B, X, Y, or Z
Description: If refPositionsFile is a PDB file that contains all the atoms in the topology, this option may be provided to set which PDB field is used to flag the reference coordinates for atoms.
-
refPositionsColValue
Atom selection flag in the PDB column
Context: rmsd
Acceptable values: positive decimal
Description: If defined, this value identifies in the PDB column
refPositionsCol of the file refPositionsFile
which atom positions are to be read. Otherwise, all positions
with a non-zero value are read.
-
atomPermutation
Alternate ordering of atoms for RMSD computation
Context: rmsd
Acceptable values: List of atom numbers
Description: If defined, this parameter defines a re-ordering (permutation) of the 1-based atom numbers that
can be used to compute the RMSD, typically due to molecular symmetry.
This parameter can be specified multiple times, each one defining a new permutation:
the returned RMSD value is the minimum over the set of permutations.
For example, if the atoms making up the group are 6, 7, 8, 9, and atoms 7, 8, and 9
are invariant by circular permutation (as the hydrogens in a CH3 group), a
symmetry-adapted RMSD would be obtained by adding:
atomPermutation 6 8 9 7
atomPermutation 6 9 7 8
Note that this does not affect the least-squares roto-translational fit,
which is done using the topology ordering of atoms, and the reference
positions in the order provided.
Therefore, this feature is mostly useful when using custom fitting parameters within the
atom group, such as fittingGroup, or when fitting
is disabled altogether.
This component returns a positive real number (in Å).
In the standard usage as described above, the rmsd component
calculates a minimum RMSD, that is, current coordinates are optimally
fitted onto the same reference coordinates that are used to
compute the RMSD value. The fit itself is handled by the atom group
object, whose parameters are automatically set by the rmsd
component.
For very specific applications, however, it may be
useful to control the fitting process separately from the definition
of the reference coordinates, to evaluate various types of
non-minimal RMSD values. This can be achieved by setting the
related options (refPositions, etc.) explicitly in the
atom group block. This allows for the following non-standard cases:
- applying the optimal translation, but no rotation
(rotateReference off), to bias or restrain the shape and
orientation, but not the position of the atom group;
- applying the optimal rotation, but no translation
(centerReference off), to bias or restrain the shape and
position, but not the orientation of the atom group;
- disabling the application of optimal roto-translations, which
lets the RMSD component describe the deviation of atoms
from fixed positions in the laboratory frame: this allows for custom
positional restraints within the Colvars module;
- fitting the atomic positions to different reference coordinates
than those used in the RMSD calculation itself
(by specifying refPositions or refPositionsFile
within the atom group as well as within the rmsd block);
- applying the optimal rotation and/or translation from a separate
atom group, defined through fittingGroup:
the RMSD then reflects the deviation from reference coordinates in a separate, moving
reference frame (see example in the section on fittingGroup).
The block eigenvector {...} defines the projection of the coordinates
of a group of atoms (or more precisely, their deviations from the
reference coordinates) onto a vector in
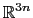 , where
, where 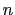 is the
number of atoms in the group. The computed quantity is the
total projection:
is the
number of atoms in the group. The computed quantity is the
total projection:
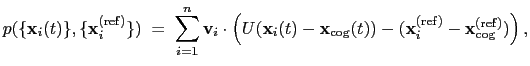 |
(13.6) |
where, as in the rmsd component, 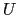 is the optimal rotation
matrix,
and
are the centers of
geometry of the current and reference positions respectively, and
is the optimal rotation
matrix,
and
are the centers of
geometry of the current and reference positions respectively, and
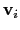 are the components of the vector for each atom.
Example choices for
are the components of the vector for each atom.
Example choices for
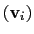 are an eigenvector
of the covariance matrix (essential mode), or a normal
mode of the system. It is assumed that
are an eigenvector
of the covariance matrix (essential mode), or a normal
mode of the system. It is assumed that
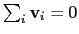 :
otherwise, the Colvars module centers the
automatically when reading them from the configuration.
:
otherwise, the Colvars module centers the
automatically when reading them from the configuration.
List of keywords (see also for additional options):
-
atoms: see definition of atoms (rmsd component)
-
refPositions: see definition of refPositions (rmsd component)
-
refPositionsFile: see definition of refPositionsFile (rmsd component)
-
refPositionsCol: see definition of refPositionsCol (rmsd component)
-
refPositionsColValue: see definition of refPositionsColValue (rmsd component)
-
vector
Vector components
Context: eigenvector
Acceptable values: space-separated list of (x, y, z) triplets
Description: This option (mutually exclusive with vectorFile) sets the values of the vector components.
-
vectorFile
file containing vector components
Context: eigenvector
Acceptable values: UNIX filename
Description: This option (mutually exclusive with vector) sets the name of a coordinate file containing the vector components; the file is read according to the same format used for refPositionsFile.
For a PDB file specifically, the components are read from the X, Y and Z fields.
Note: The PDB file has limited precision and fixed-point numbers: in some cases, the vector components may not be accurately represented; a XYZ file should be used instead, containing floating-point numbers.
-
vectorCol
PDB column used to flag participating atoms
Context: eigenvector
Acceptable values: O or B
Description: Analogous to atomsCol.
-
vectorColValue
Value used to flag participating atoms in the PDB file
Context: eigenvector
Acceptable values: positive decimal
Description: Analogous to atomsColValue.
-
differenceVector
The
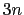 -dimensional vector is the difference between vector and refPositions
-dimensional vector is the difference between vector and refPositions
Context: eigenvector
Acceptable values: boolean
Default value: off
Description: If this option is on, the numbers provided by vector or vectorFile are interpreted as another set of positions,
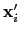 : the vector
is then defined as
: the vector
is then defined as
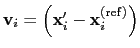 .
This allows to conveniently define a colvar
.
This allows to conveniently define a colvar 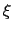 as a projection on the linear transformation between two sets of positions, ``A'' and ``B''.
For convenience, the vector is also normalized so that
as a projection on the linear transformation between two sets of positions, ``A'' and ``B''.
For convenience, the vector is also normalized so that 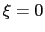 when the atoms are at the set of positions ``A'' and
when the atoms are at the set of positions ``A'' and 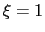 at the set of positions ``B''.
at the set of positions ``B''.
This component returns a number (in Å), whose value ranges between
the smallest and largest absolute positions in the unit cell during
the simulations (see also distanceZ). Due to the
normalization in eq. 13.6, this range does not
depend on the number of atoms involved.
The block gyration {...} defines the
parameters for calculating the radius of gyration of a group of atomic
positions
with respect to their center of geometry,
:
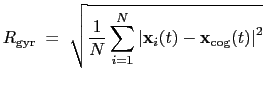 |
(13.7) |
This component must contain one atoms {...} block to
define the atom group, and returns a positive number, expressed in
Å.
List of keywords (see also for additional options):
-
atoms: see definition of atoms (rmsd component)
The block inertia {...} defines the
parameters for calculating the total moment of inertia of a group of atomic
positions
with respect to their center of geometry,
:
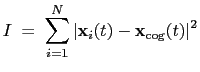 |
(13.8) |
Note that all atomic masses are set to 1 for simplicity.
This component must contain one atoms {...} block to
define the atom group, and returns a positive number, expressed in
Å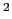 .
.
List of keywords (see also for additional options):
-
atoms: see definition of atoms (rmsd component)
The dipoleMagnitude {...} block defines the dipole magnitude of a group of atoms (norm of the dipole moment's vector), being atoms the group where dipole magnitude is calculated.
It returns the magnitude in elementary charge 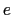 times Å.
times Å.
List of keywords (see also for additional options):
-
atoms: see definition of atoms (rmsd component)
The block inertiaZ {...} defines the
parameters for calculating the component along the axis
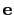 of the moment of inertia of a group of atomic
positions
with respect to their center of geometry,
:
of the moment of inertia of a group of atomic
positions
with respect to their center of geometry,
:
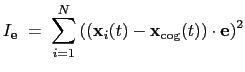 |
(13.9) |
Note that all atomic masses are set to 1 for simplicity.
This component must contain one atoms {...} block to
define the atom group, and returns a positive number, expressed in
Å
.
List of keywords (see also for additional options):
-
atoms: see definition of atoms (rmsd component)
-
axis
Projection axis (Å)
Context: inertiaZ
Acceptable values: (x, y, z) triplet
Default value: (0.0, 0.0, 1.0)
Description: The three components of this vector define (when normalized) the
projection axis
.
Next: Rotations
Up: Defining collective variables
Previous: Contacts
Contents
Index
vmd@ks.uiuc.edu