



Next: Collective metrics
Up: Defining collective variables
Previous: Angles
Contents
Index
Subsections
The coordNum {...} block defines
a coordination number (or number of contacts), which calculates the
function
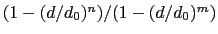 , where
, where 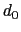 is the
``cutoff'' distance, and
is the
``cutoff'' distance, and 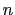 and
and 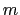 are exponents that can control
its long range behavior and stiffness [49]. This
function is summed over all pairs of atoms in group1 and
group2:
are exponents that can control
its long range behavior and stiffness [49]. This
function is summed over all pairs of atoms in group1 and
group2:
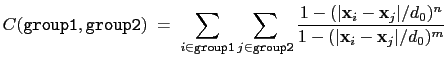 |
(13.3) |
List of keywords (see also ![[*]](crossref.png) for additional options):
for additional options):
-
group1: see definition of group1 (distance component)
-
group2: analogous to group1
-
cutoff
 ``Interaction'' distance (Å)
``Interaction'' distance (Å)
Context: coordNum
Acceptable values: positive decimal
Default value: 4.0
Description: This number defines the switching distance to define an
interatomic contact: for 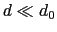 , the switching function
is close to 1, at
, the switching function
is close to 1, at 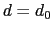 it
has a value of
it
has a value of 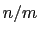 (
(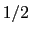 with the default
and
), and at
with the default
and
), and at
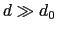 it goes to zero approximately like
it goes to zero approximately like 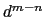 . Hence,
for a proper behavior,
must be larger than
.
. Hence,
for a proper behavior,
must be larger than
.
-
cutoff3
Reference distance vector (Å)
Context: coordNum
Acceptable values: ``(x, y, z)'' triplet of positive decimals
Default value: (4.0, 4.0, 4.0)
Description: The three components of this vector define three different cutoffs
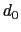 for each direction. This option is mutually exclusive with
cutoff.
for each direction. This option is mutually exclusive with
cutoff.
-
expNumer
Numerator exponent
Context: coordNum
Acceptable values: positive even integer
Default value: 6
Description: This number defines the
exponent for the switching function.
-
expDenom
Denominator exponent
Context: coordNum
Acceptable values: positive even integer
Default value: 12
Description: This number defines the
exponent for the switching function.
-
group2CenterOnly
Use only group2's center of
mass
Context: coordNum
Acceptable values: boolean
Default value: off
Description: If this option is on, only contacts between each atoms in group1 and the center of mass of group2 are calculated (by default, the sum extends over all pairs of atoms in group1 and group2).
If group2 is a dummyAtom, this option is set to yes by default.
-
tolerance
Pairlist control
Context: coordNum
Acceptable values: decimal
Default value: 0.0
Description: This controls the pairlist feature, dictating the minimum value for each summation element in Eq. 13.3 such that the pair that contributed the summation element is included in subsequent simulation timesteps until the next pairlist recalculation. For most applications, this value should be small (eg. 0.001) to avoid missing important contributions to the overall sum. Higher values will improve performance by reducing the number of pairs that contribute to the sum. Values above 1 will exclude all possible pair interactions. Similarly, values below 0 will never exclude a pair from consideration. To ensure continuous forces, Eq. 13.3 is further modified by subtracting the tolerance and then rescaling so that each pair covers the range
![$ \left[0, 1\right]$](img172.png) .
.
-
pairListFrequency
Pairlist regeneration frequency
Context: coordNum
Acceptable values: positive integer
Default value: 100
Description: This controls the pairlist feature, dictating how many steps are taken between regenerating pairlists if the tolerance is greater than 0.
This component returns a dimensionless number, which ranges from
approximately 0 (all interatomic distances are much larger than the
cutoff) to
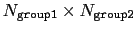 (all distances
are less than the cutoff), or
(all distances
are less than the cutoff), or
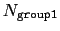 if
group2CenterOnly is used. For performance reasons, at least
one of group1 and group2 should be of limited size or group2CenterOnly should be used: the cost of the loop over all pairs grows as
.
Setting
if
group2CenterOnly is used. For performance reasons, at least
one of group1 and group2 should be of limited size or group2CenterOnly should be used: the cost of the loop over all pairs grows as
.
Setting
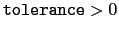 ameliorates this to some degree, although every pair is still checked to regenerate the pairlist.
ameliorates this to some degree, although every pair is still checked to regenerate the pairlist.
The selfCoordNum {...} block defines
a coordination number similarly to the component coordNum,
but the function is summed over atom pairs within group1:
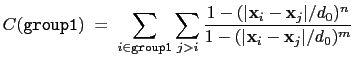 |
(13.4) |
The keywords accepted by selfCoordNum are a subset of
those accepted by coordNum, namely group1
(here defining all of the atoms to be considered),
cutoff, expNumer, and expDenom.
List of keywords (see also for additional options):
-
group1: see definition of group1 (coordNum component)
-
cutoff: see definition of cutoff (coordNum component)
-
cutoff3: see definition of cutoff3 (coordNum component)
-
expNumer: see definition of expNumer (coordNum component)
-
expDenom: see definition of expDenom (coordNum component)
-
tolerance: see definition of tolerance (coordNum component)
-
pairListFrequency: see definition of pairListFrequency (coordNum component)
This component returns a dimensionless number, which ranges from
approximately 0 (all interatomic distances much larger than the
cutoff) to
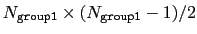 (all
distances within the cutoff). For performance reasons,
group1 should be of limited size, because the cost of the
loop over all pairs grows as
(all
distances within the cutoff). For performance reasons,
group1 should be of limited size, because the cost of the
loop over all pairs grows as
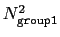 .
.
The hBond {...} block defines a hydrogen
bond, implemented as a coordination number (eq. 13.3)
between the donor and the acceptor atoms. Therefore, it accepts the
same options cutoff (with a different default value of
3.3 Å), expNumer (with a default value of 6) and
expDenom (with a default value of 8). Unlike
coordNum, it requires two atom numbers, acceptor and
donor, to be defined. It returns an adimensional number,
with values between 0 (acceptor and donor far outside the cutoff
distance) and 1 (acceptor and donor much closer than the cutoff).
List of keywords (see also for additional options):
-
acceptor
Number of the acceptor atom
Context: hBond
Acceptable values: positive integer
Description: Number that uses the same convention as atomNumbers.
-
donor: analogous to acceptor
-
cutoff: see definition of cutoff (coordNum component)
Note: default value is 3.3 Å.
-
expNumer: see definition of expNumer (coordNum component)
Note: default value is 6.
-
expDenom: see definition of expDenom (coordNum component)
Note: default value is 8.
Next: Collective metrics
Up: Defining collective variables
Previous: Angles
Contents
Index
vmd@ks.uiuc.edu