


Next: Remote connection objects
Up: Function and Class
Previous: Display objects
The following objects are used to store and represent the molecules
in VMD, including their static data as well as their dynamic data.
Items which have a specific section listed for them are
explained in detail in that section.
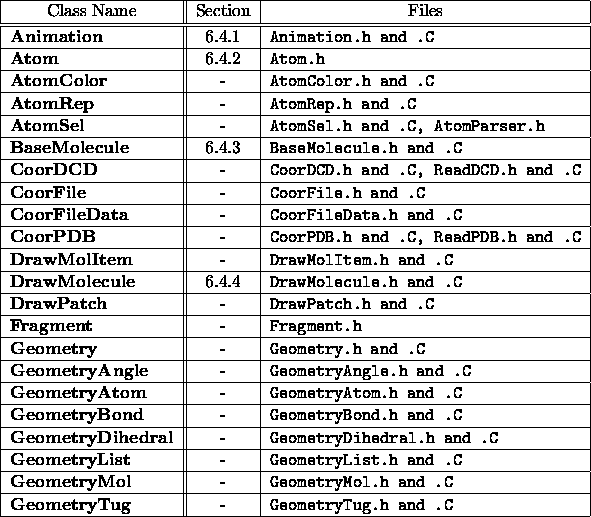
Table 6.3: VMD
molecule objects, part 1.
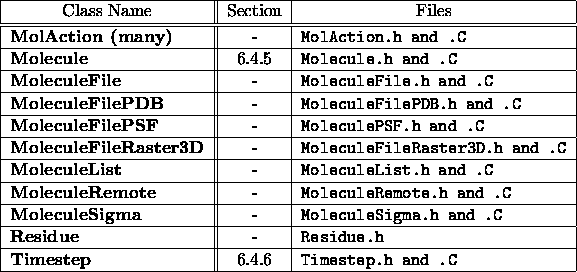
Table 6.4: VMD
molecule objects, part 2.

Animation
is a base class for all Molecule
objects in VMD
.
It is responsible for storing the dynamic data for molecule -- those
items which vary with time, instead of remaining constant. Animation
stores an animation list, and has controls to add new data to the
end of the list, as well as to position a current frame pointer in
the list. Each time a Molecule
is prepared (by calling it's
virtual prepare routine since Molecule
is derived from
Displayable
), the current position in the animation list is updated.
The Animation
consists of basically an array of Timestep
objects. Each Timestep
contains the data for the molecule for a
single step in the trajectory of the system.
The AnimDir enumeration lists the different directions in which the
animation can be moving:
- FORWARD: Animation proceeds continually forward each update.
- FORWARD1: Animation goes to the next step, then is PAUSEd.
- REVERSE: Animation proceeds continually backward each update.
- REVERSE1: Animation goes to the prev step, then is PAUSEd.
- PAUSE: Updates to not change the current position in the list.
The AnimPos enumeration lists different positions in the list to which
the animation position can jump, or can have new elements inserted or deleted:
- END: The end of the animation list.
- START: The start of the animation list.
- BEFORE: The frame before a specified frame.
- AFTER: The frame after a specified frame.
- DELETE: Unused.
- WRITE: Unused.
The AnimFrames enumeration lists the different ways in which frames
can be added or deleted. It is either ALL, or SELECTION.
The AnimStyle enumeration lists the different ways in which animation
can be done:
- ONCE: When the animation gets to the end or beginning, it will
then pause.
- LOOP: When the animation gets to the end or beginning, loop
around to the other end and continue through again in the same direction.
- ROCK: When the animation gets to the end or beginning, reverse
direction and continue animating.
- double lastTime -- the clock time when the last position was
determined. This is used to determine if the animation should proceed to
a new frame, to control the speed.
- int frameChanged -- whether the animation has changed to a
new frame recently.
- int needSetFrameChanged -- when this flag is set, then the
frameChanged flag should be changed the next time it is possible to do
so.
- ResizeArray<Timestep *> tsList -- the frames themselves.
- int currFrame -- the current frame, an index into tsList.
- int frameSkip -- if we are jumping to a new frame, this
indicates the frame to jump to.
- float Speed -- the animation speed, from 0 (slowest) to 1
(fastest).
- AnimPos appendPos -- the method to use the next time a frame
is to be appended.
- int appendFrame -- if we are inserting frames, the frame which
we are to append before or after.
- AnimDir animDir -- the current animation direction.
- AnimStyle animStyle -- the method for doing animation.
- int moveTo -- if this is not negative, this indicates a frame
to which we are to jump the next time the animation position is updated.
- float currTime -- the time which has elapsed since the
beginning of the animation.
- int num(void) -- returns current number of frames in the list.
- int frame(void) -- returns the current frame number.
- int is_current(void) -- returns whether there is a
current frame.
- Timestep *current(void) -- returns the current frame itself,
or NULL if there is none.
- Timestep *item(int) -- returns the Nth frame.
- void goto_frame(int) -- jump to the specified frame.
- int has_frame_changed(void) -- returns whether the current
frame has been changed recently.
- void delete_animation(void) -- deletes all the currently
stored frames.
- void delete_frame(int) -- delete the Nth frame.
- int append_frame(Timestep *) -- adds the given frame to the
end of the animation list (or wherever has been previously determined for it
to be placed).
- int anim_update(void) -- updates the current position of the
animation in the list, based on the current animation mode, and returns the
index of the new current frame. Also sets the flags indicating whether the
frame has changed. This is called during each call to prepare in the
Molecule
object.
- void append_end(void) -- indicate to append the next frame at
the end of the list.
- void append_start(void) -- indicate to append the next frame at
the start of the list.
- void append_after(int) -- indicate to append the next frame
after the specified frame.
- void append_before(int) -- indicate to append the next frame
before the specified frame.
- void skip(int) -- set the current frame skip rate (the increment
by which the current frame pointer is changed).
- int skip(void) -- return the current frame skip rate.
- void anim_dir(AnimDir) -- set the current animation direction.
- AnimDir anim_dir(void) -- return the animation direction.
- void anim_style(AnimStyle) -- set the animation method.
- AnimSTyle anim_style(void) -- return the animation method.
- float speed(float) -- set the speed of the animation.
- float speed(void) -- return the current animation speed.
To add new frames, do the following:
- Set the method for appending, either at the end, or beginning, or
before or after some current frame.
- Call the append routine.
The anim_update routine
changes the current position based on the current animation direction
and style. It also sets flags indicating whether the current frame was
changed any. It will return the index of the new current frame, which
can be used to get the actual Timestep
object for that frame.
Right now, this is very top-level base class for Molecule
, even higher
than the BaseMolecule
level. The functionality here could probably
be rolled into BaseMolecule
, so that a single class would have info
about the structure and size of the molecule is was storing dynamic data for.
Basically, it's backwards to have Animate
above BaseMolecule
.

An Atom
object stores the data for a single atom in a molecule. Each
atom has the following information:
- Mass
- Charge
- Atom name
- Atom type
- Residue name
- Residue ID number
- Chain identifier
- Segment name
- List of bonds
Each BaseMolecule
contains a list of these Atom
objects.
- Atom::(int, float *, float *, char *, char *, char *,
char *, char *, char *)
The first argument is the index of the atom; the rest are the names used
to distinguish this atom (as mentioned in the list above).
Each atom has a set of `extra' data values, other than it's position. The
codes used as indices for these extra data values are:
- ATOMCHARGE
- ATOMMASS
- ATOMBETA
- ATOMOCCUP
- ATOMRAD
- ATOMEXTRA -- the number of `extra' data items.
The BackboneType enumeration lists the different types of backbone
bonds which may exist:
- NORMAL -- used if nothing else special about the backbone
bond can be determined.
- PROTEINBACK -- used for protein backbone bonds.
- NUCLEICBACK -- used for nucleic acid backbone bonds.
The ResidueType enumeration lists the different classes of residues
which this atom may be a part of:
- NOTHING -- not in a residue of any noted special group.
- PROTEIN -- a standard protein residue, i.e. an amino acid.
- NUCLEIC -- a standard nucleic acid resiude, i.e. A,T,G,C, or U.
- WATERS -- a solvent residue, most likely water.
- int index -- the index of this atom, as provided in the
constructor.
- int bonds -- the total number of bonds which this atoms
participates in. Note that a bond will be stored twice, once for each of
the atoms it connects.
- int uniq_resid -- a unique integer indicating which residue
this atom is in. This is not the same as te residue ID, since that is not
necessarily unique in a given structure (particularly if the structure
contains more than one segment or chain).
- int fragment -- which fragment this atom belongs in.
- int bondTo[MAXATOMBONDS] -- the indices of the other atoms to
which this atom is bonded. An atom can bond to a maximum number of other
atoms before an error occurs, this maximum is currently 8.
- BackboneType bondType[MAXATOMBONDS] -- lists what type of bond
each to the bonds to this atom are.
- BackboneType atomType -- indicates if this atom is part of the
backbone, and if so what kind of backbone.
- ResidueType residueType -- what type of residue this atom is
a part of.
- float pos[], extra[] -- position and `extra' data values for
this atom. These are initial, or `default', values for the atom when no
other coordinates or extra values are known.
- char namestr[], int nameindex -- the atom name, and the index
of that name in the unique atom name array in the BaseMolecule
which
created the Atom
object.
- char typestr[], int typeindex -- the atom type name, and the
index of that name in the unique atomtype name array in the BaseMolecule
which created the Atom
object.
- char resnamestr[], int resnameindex -- the residue name, and the
index of that name in the unique residue name array in the BaseMolecule
which created the Atom
object.
- char residstr[], int residindex -- the residue ID, and the
index of that name in the unique residue ID array in the BaseMolecule
which created the Atom
object.
- char segnamestr[], int segnameindex -- the segment name,
and the index
of that name in the unique segment name array in the BaseMolecule
which
created the Atom
object.
- char chainstr[], int chainindex -- the chain ID,
and the index
of that ID in the unique chain ID array in the BaseMolecule
which
created the Atom
object.
- float radius() -- return the atom radius.
- float mass() -- return the atom mass.
- float charge() -- return the atom charge.
- float beta() -- return the atom beta value.
- float occup() -- return the atom occupancy value.
- void add_bond(int, BackboneType) -- add a new bond to this
atom, which connects this atom and the specified atom, of the given
bond type.
- int bonded(int a) -- return TRUE if this atom is bonded
to the specified atom.
To create a new Atom
, you specify all the names in the constructor,
and then add the bonds to the atom one at a time using the add_bond
routine. Later, when the molecular structure has been analyzed, the
flags indicating the residue type, fragment index, etc. must be set (they
are currently public members, so they can be accessed directly).
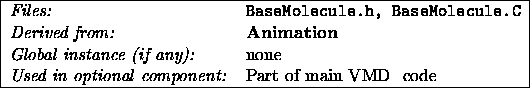
BaseMolecule
is the second-highest level in the class hierarchy for
Molecule
objects; it is derived from Animation
, and is used
to store the static information for the molecule, which is the
basic information about the structure and contents of the system which do
not change with time. BaseMolecule
has no ability to read in this
information itself, instead it provides routines which derived classes call
in order to add new molecules, bonds, etc. to the molecule.
BaseMolecule
has no responsibility for graphically displaying the
molecule, either; that is left to the DrawMolecule
class, which is
derived from BaseMolecule
and Displayable
.
You do not ever create a BaseMolecule
instance directly, instead you
create an instance of a class derived from Molecule
, for which
BaseMolecule
is a base class (among others). When initially created,
a new BaseMolecule
is empty, with zero atoms and zero bonds. The
derived classes contain the actual code to read in the molecular structure
from a file or from a network connection, and they add the components to
the internal storage via routines in BaseMolecule
. When all the
structure is completely read in, then routine are called in BaseMolecule
to analyze the structure, and calculate such things as what atoms are in
what residues, how many residues there are, what are the backbone bonds,
etc. In fact, a molecule contains these structural features which are
either directly added to BaseMolecule
, or calculated by
BaseMolecule
after the basic structure is read in:
- N atoms, which are added to the system as they are read in from some source. Each atom has associated with it several names, which help to
distinguish the atoms and make it possible for the VMD
atom selection
mechanism to choose subsets of atoms. These names are:
- The atom name, which is usually a standard chemical
nomenclature name. For example, alpha carbons in proteins have the name
CA.
- The atom type, which for some molecular data files is a
name from a much smaller set of total names, used to classify atoms into
small sets which are more manageable. For example, PSF files use atom types
to simplify the parameterization of the atoms for molecular dynamics
simulations. If the atom type is not known from the input files, it is just
set to be the same as the atom name.
- The residue name, a three-letter code which is usually
quite standard. All glycine amino acids, for example, are in residues with
the name GLY.
- The residue ID, a numeric value assigned to the residues in
a molecule, quite often in increasing order from one end of a linear chain
to another. Particular useful in proteins, which are unbranched polymers.
- The chain ID, a single-letter code used to distinguish atoms
among different subcomponents of a molecule. If it is unknown for an atom,
it is given a default value of `X'.
- The segment name, similar to the chain ID but allowed to be
up to four characters. This is not as standard as the chain ID, and if it
is unknown it will be given a default value of MAIN.
- N bonds, which are added to the system as they are read in or
calculated by a derived class. The bonds are not stored directly in a list,
however; instead, each atom stores a list of the bonds it participates in,
which makes it much faster to display the molecule.
- N protein backbone bonds, and N nucleic-acid backbone bonds,
which are determined by BaseMolecule
after the atoms and bonds are
added.
- N residues, where each residue is a collection of atoms and bonds
which form some subunit.
- N segments, which each consist of a collection of atoms in a
functional substructure within the molecule. For example, quite often a
protein is a segment, and surrounding water molecules are another segment.
- N fragments, where each fragment is a collection of connected
residues. If a system consists of three disconnected alpha helices, for
example, then each helix would be a separate fragment. There are lists of
protein fragments as well as nucleic acid fragments.
After the atoms and bonds are added to the BaseMolecule
, then the
connectivity is analyzed and the names of the atoms are used to find the
backbone bonds, the residues in the system, and the fragments. Atoms, bonds,
residues, and other components are numbered 0 ... N-1 in their respective
lists.
One other item which BaseMolecule
stores is the unique molecule ID
number, which is assigned when the molecule is created. Each new
molecule in VMD
gets assigned an integer ID. The assigned ID values
increase by one as each new system is loaded. The commands used to affect
the molecules use these ID numbers to determine which molecule the command
should affect. The name of the molecule displayed in the Molecule
on-screen menu form has this ID number appended to the end of it.
The MoleculeType enumeration lists the different type of molecules
which VMD
understands. When the structure is analyzed, the type of
molecule is determined. The types are:
- UAPROTEIN
- EHPROTEIN
- UAPROTDNA
- EHPROTDNA
- NUCLEIC
- ORGANIC
- INORGANIC
- MoleculeType type -- type of this molecule (from above list).
- int nAtoms -- number of atoms in this molecule. Can be zero.
- int nBonds -- number of bonds in this molecule.
- int nBackProtein -- number of protein backbone bonds.
- int nBackDNA -- number of nucleic-acid backbone bonds.
- int nResidues -- number of residues.
- int nSegments -- number of segments.
- int ID -- molecule integer ID number.
- int maxAtoms -- maximum storage currently allotted to store the
atoms (i.e. size of atomList array, which may be larger than the
actual number of atoms stored there).
- Atom **atomList -- array of Atom
objects.
- NameList<int> atomNames -- list of unique atom names in this
molecule.
- NameList<int> atomTypes -- list of unique atom types in this
molecule.
- NameList<int> resNames -- list of unique residue names in this
molecule.
- NameList<int> resIds -- list of unique residue ID's in this
molecule.
- NameList<int> chainNames -- list of unique chain ID's in this
molecule.
- NameList<int> segNames -- list of unique segment names in this
molecule.
- ResizeArray<Residue *> residueList -- list of which residues
are connected to which.
- ResizeArray<Fragment *> fragList -- list of connected residues,
which form fragments.
- ResizeArray<Fragment *> pfragList -- list of connected protein
residues which form protein fragments. A protein fragment is a single chain
from N to C.
- ResizeArray<Fragment *> nfragList -- list of connected nucleic
acid residues, which form nucleic acid fragments. A nucleic acid fragment
is a single chain from 5' to 3'.
- void init_atoms(int) -- initializes storage to store data
for N atoms. This only allocates memory, it does not store anything in that
memory. This should be called when constructing a new molecule, when the
number of atoms has been determined but before the atom data itself is
stored into the BaseMolecule
structures.
- int add_atom(char *, char *, char *, char *,
char *, char *, float *pos, float *extra) -- add a new atom to
the molecule, with the specified names, and given starting x, y, z position
( pos) and given starting extra data (such as beta value and occupancy).
- int add_bond(int, int, Atom::BackboneType = Atom::NORMAL) --
add a new bond between the atoms specified as the first two arguments, where
the bond is of the specified type. See the description of Atom
for a
list of the different bond types.
- int find_backbone(void) -- determines which bonds are backbone
bonds, and stores this data in the Atom
objects stored in the
atomList member. Returns the number of backbone bonds found.
- int find_residues(void) -- find which atoms are in which
residues, and store this data. Returns the number of residues found.
- int find_waters(void) -- Find the waters, based on their
residue name, and return the number found.
- int find_segments(void) -- Find the segments in the molecule,
and store this data. Return the number round.
- int find_fragments(void) -- Find the fragments in the molecule,
and store this data. Return the number found.
- int find_atom_in_residue(char *nm, int r) -- find the index
of the first atom in the specified residue with the given name, or return
-1 if none is found with that name.
- int id(void) -- return the ID of the molecule.
- Atom *atom(int) -- return the Nth Atom
for the molecule.
- char *atom_full_name(int, char * = NULL) -- return a string
containing the full name specification for the Nth atom. If the second
argument is not NULL, the name will be placed in the given character array.
Otherwise, an internal static buffer will be used to hold the name. The name
is of the form:
<mol ID>:<atom index>
This name is guaranteed to be unique for each atom.
- char *atom_short_name(int, char * = NULL) -- the same as
for the full name, except the name returned is of the form:
<residue name><residue ID>:<atom name>
This form is nicer to read, but is not generally unique for a given atom.
- float default_charge(char *) -- returns a default partial
charge to use for the specified atom name. Used when this information is
not supplied by the source of molecular structure. The following routines
also supply default data based on a given atom name.
- float default_mass(char *)
- float default_radius(char *)
- float default_occup(char *)
- float default_beta(char *)
- virtual int create(void) -- the main virtual routine provided
by this class. This is used after a new Molecule
subclass has been
created (with the required information for reading the molecule given in
the constructor). Initially the Molecule
is empty; to initialize it,
the create() routine is called which will then start the actual
process of reading in the data. Each version of create() supplied
by the derived classes should, after doing it's own creation, call the
create() routine in the parent class. This routine returns the
success of the creation operation.
- virtual float scale_factor(void) -- returns (possibly
calculating first) the scaling factor required to scale the coordinates for
the current timestep to fit in a box from -1 ... 1 in all dimensions.
- virtual void cov(float &, float &, float &) -- return
the position of the center of volume of the current coordinate set.
A new molecule is first created by using `new' with the proper subclass of
Molecule
( Molecule
is the `standard' class to use for all
molecule objects in VMD
; classes derived from Molecule
are specialized
to read in data from different sources, while classes above the Molecule
level only deal with some of the information required to store and display
and animate a structure.). Then, after the new instance is assigned to
a Molecule
pointer, then the create() virtual function should
be called. This will actually result in all the action being done, for
example data files will be read or network connections will be established.
The version of create() in BaseMolecule
should be called
after the molecule has been read in by the derived classes. It
analyzes the structure and finds the backbone bonds, fragments, etc. When
create() is finished, the molecule is ready to go. If create()
does not return TRUE, however, the creation failed (i.e. the files could
not be opened), and the new molecule will still be empty.
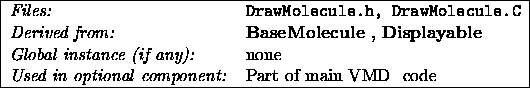
DrawMolecule
is derived from BaseMolecule
, and is the next
level in the Molecule
class hierarchy. It embodies the level of
control and functionality responsible for creating a graphical image of
the molecule. It is derived from Displayable
(actually, from
Displayable3D), and so will be part of the Scene
that will
be drawn by a DisplayDevice
.
Each DrawMolecule
keeps a list of DrawMolItem
objects; each
DrawMolItem
is also a Displayable
, and is responsible for a
single representation of the molecule. Each representation contains
a display list used to draw a single view of the molecule. To define a
single representation, each DrawMolItem
contains one instance each
of the following objects:
- AtomColor
-- defines how to color each atom in the representation.
- AtomRep
-- defines what shape to draw the components of the
representation.
- AtomSel
-- determines which atoms out of all the ones in the
molecule are to be drawn in that representation.
When a new DrawMolecule
is created (i.e. when a new Molecule
is created), it is added as a child Displayable
to a global
MoleculeList
object. Thus, you can rotate just the MoleculeList
,
and this will rotate all the molecules at the same time. When a molecule
is first displayed, it is scaled and translated to fit within a -1 ... 1
size box centered around the origin. In order to have all the molecules be
the proper size in relation to each other, and to preserve the spatial
position of the molecules, only one molecule is used to determine the
scaling and translation positions, and the others then use those same
scaling and translation values.
Each DrawMolItem
added to this object are added as child
Displayable
objects of DrawMolecule
, and so scaling and rotating
the molecule will do so to all the DrawMolItem
objects at the same
time.
- int active -- is this molecule an active one? This is used
by MoleculeList
.
- int needRedraw -- a flag indicating whether this item needs
to reconstruct its display list during the prepare routine. After the
display list is remade, this flag is cleared.
- ResizeArray<DrawMolItem *> repList -- the list of
representations.
- int atom_displayed(int) -- returns TRUE if any of the
representations are currently displaying the given atom.
- int components(void) -- returns the number of representations
in the molecule.
- DrawMolItem *component(int) -- return a pointer to the Nth
representation.
- int del_rep(int) -- delete the Nth representation, and return
the success.
- virtual void set_name(char *) -- used to change or set the
name of the molecule. Called during the create phase of molecule
construction.
- virtual void create_cmdlist(void) -- regenerates the
display list, which contains the list of primitive drawing commands
necessary to draw the object.
- virtual int create(void) -- the version of create for
DrawMolecule
... this is called by derived classes after they have
read in and initialized the molecule. The DrawMolecule
version of
create will then call the BaseMolecule
's create routine, and finally
then construct the initial version of the display list.
- virtual void prepare(DisplayDevice *) -- provided since this
is a Displayable
object. This determines if the display list needs
to be reconstructed, and if so it does the reconstruction.
- virtual int add_rep(AtomColor *, AtomRep *, AtomSel *) --
requests for a new representation to be made, using the given objects to
describe what the representation should be. Returns success.
- virtual int change_rep(int, AtomColor *, AtomRep *, AtomSel *)
-- changes the Nth representation to use the new settings specified in the
given objects.
The user should never create a DrawMolecule
directly; it should instead
be used as a base class for Molecule
.

This is the class used generally throughout VMD
to represent and access
a molecule. It is derived from several base classes which provide the
storage and control over the molecule's structure ( BaseMolecule
),
animation list ( Animation
), and graphical display ( DrawMolecule
).
It still does not contain the abilities to read in new molecular data from
different sources, instead these are in classes derived from Molecule
.
This class is used as the basic class that all other parts of VMD
\
are aware of, since once a molecule is read in from some source, it acts
just like any other molecule.
The one level of extra functionality which is provided directly by the
Molecule
class is the ability to read in new coordinate frames from
an external coordinate file, either a PDB or a DCD file.
- Molecule::(char *, Scene *)
The first argument is the source of the data, it is a string
indicating either a file, or some remote computer, or perhaps some other
source. The second argument is the Scene
to which this molecule
should be added. This version of the constructor should only be used if
no MoleculeList
is being used.
- Molecule::(char *, Displayable *)
Same as above, except a parent Displayable
is specified instead of
a Scene
. This is the version that should be used mainly in VMD
,
with the global MoleculeList
object specified as the parent
Displayable
for the molecule.
- char *source -- the source of the molecule structure data.
- ResizeArray<CoorFileData *> coorIOFiles -- data on which files
containing atomic coordinates are to be read in by this object. Each
CoorFileData
instance contains data on which file to read, what type
of file it is, and which frames to read from the file. The same data
is stored in the case where a file is to be written instead of read.
- int read_coor_file(char *, int, int=(-1), int=(-1), int=(-1))
-- request to read in the specified file. The second argument is the type,
and the remaining are the beginning frame, ending frame, and frame skip
values. If any are not specified or are < 0, default values are used.
- write_coor_file(char *, int, int=(-1), int=(-1), int=(-1))
-- same as above, but requests to write out a file.
- virtual void prepare(DisplayDevice *) -- does any preparation
necessary before redrawing. If data files are being read, this will read
the next frame from them.
New molecules are created in VMD
in the CmdMolNew
object, which is
a particular Command
-derived object. When a new molecule is to be
created, the type of molecule source and necessary data are specified (such
as filenames, remote computer names, etc) and given to the constructor of
a specialized subclass of Molecule
. The resulting (empty) molecule
is assigned to a Molecule * pointer variable, and then the virtual
create routine is called. After this, the molecule is added to
the MoleculeList
, and any extra actions are taken such as requesting
for more coordinate files to be read, etc.

The Timestep
class is used to hold the dynamic data for a Molecule
for a single step in a trajectory. It hold the coordinates, velocities,
energies, etc. that vary with time. As each new Timestep
is read in
from some source (a file, or from a network connection) it is added to
the animation list for the associated molecule; this list is maintained
by the Animation
object, from which each Molecule
is derived.
Almost all the items in this class are public, so they may be retrieved
quickly by other objects in VMD
. This is not such a great design, however;
see the notes below about future changes.
- Timestep::(int n, float DT, float *newdata = NULL)
The first argument is the number of objects (atoms) for which dynamic data
will be stored. The second is the time which has elapsed between generation
of this step a previous step (for example, for a molecular dynamics
simulation, this would be the integration timestep). The final argument is
the data to store in this step, if it has already been allocated. If not,
new space will be allocated by Timestep
.
There are a number of globally defined macros in this class which are used
as indices into the data arrays stored by this object. They are:
- Energy Data Indices:
- TSE_BOND -- index to bond energy value.
- TSE_ANGLE -- index to angle energy value.
- TSE_DIHE -- index to dihedral energy value.
- TSE_IMPR -- index to improper dihedral energy value.
- TSE_VDW -- index to van der Waal energy value.
- TSE_COUL -- index to Coulomb energy value.
- TSE_HBOND -- index to hydrogen-bond energy value.
- TSE_KE -- index to total kinetic energy value.
- TSE_PE -- index to total potential energy value.
- TSE_TOTAL -- index to total energy value.
- TSE_TEMP -- index to temperature value.
- Per-Atom Data Indices:
- TSP_X -- x coordinate.
- TSP_Y -- y coordinate.
- TSP_Z -- z coordinate.
- Per-Patch Data Indices:
- TSP_XLEN -- length of patch in x direction.
- TSP_YLEN -- length of patch in y direction.
- TSP_ZLEN -- length of patch in z direction.
- TSP_LOAD -- computational load on the patch.
- TSP_ATOMS -- number of atoms in the patch.
- TSP_NODE -- node which contains the patch.
- int needDataDelete -- flag for whether we need to delete the
storage space for the data array when this object is deleted.
- int num -- number of atoms in this Timestep
.
- float *pos -- coordinates of all the atoms. This array has
3 * num elements, arranged as ([x1 y1 z1] [x2 y2 z2] ...).
- float *data -- for each atom, there can be `extra' data,
which Timestep
does not not know the contents of. This is the data
provided as the last argument in the constructor.
- float energy[] -- energy values for this timestep. By default,
they are zero unless they are explicitly changed by the creator of this
timestep.
- int numPatches -- the number of patches stored in this step.
It may be zero (and definitely is zero for data which did not come from a
network connection).
- float *patchData -- data stored for each patch (the size,
location, atoms per patch, etc.).
- float minpos[], maxpos[], mindata[], maxdata[], etc -- the
minimum and maximum values found in this Timestep
for the various
data quantities. Several parts of VMD
need to know the range of data values
some quantity takes in a Timestep
.
- float COV[] -- the x,y,z position of the center of volume.
- float scale_factor -- the amount by which you would need to
scale the coordinates in this step in order to fit them in a box in the
range (-1 ... 1) in all dimensions.
- float dt -- the time required to compute this timestep. If this
is unknown, it is set to zero.
- int Initialized -- has this been initialized? If so, then
all the data items have been placed into the proper arrays, and the max
and min values have been calculated.
- float *create_patch_storage(int) -- allocates internal storage
to store data for the given number of patches. The number of patches is
stored internally as well. Returns a pointer to the newly-allocated
array.
- void init(void) -- after all the data has been placed into a
new Timestep
instance, this routine calculate the max/min values
for all the quantities, and anything else required based on atom
coordinates, etc. Used for scaling and translating purposes.
A new Timestep
is created each time new coordinate data needs to be
added to the end of the animation list. Use the following steps:
- Create a new instance:
Timestep *ts = new Timestep(atoms, dt);
- Copy coordinate and energy data into the proper arrays, by
accessing the public data items described above.
- If there is patch data, first call create_patch_storage with
the number of patches (to allocate storage), and then copy the patch
data into the array which is returned by the function.
- After all data is entered, the last step is to call the init
routine. This goes through all the stored quantities (x, y, z position,
atoms/patch, etc) and calculates the maximum and minimum values. It also
finds the position of the center of volume, and the proper scaling factor.
This could perhaps benefit from having a NameList
store the different
quantities, with a specific name associated with each quantity. Then,
instead of having to hard-code into Timestep
what data is being stored,
you could just have a routine to add new items with a given name to the
object, and other routines to access data by providing the proper name
and index (or maybe just provide the name, and have returned the proper
array storing the data).
Next: Remote connection objects
Up: Function and Class
Previous: Display objects
Andrew Dalke
Wed May 15 02:25:03 CDT 1996