From: Robert Johnson (robertjo_at_physics.upenn.edu)
Date: Wed Jun 20 2012 - 11:30:29 CDT
Hi Jerome,
Your idea of using the RMSD sounds like a good one to me. We don't expect
to get a rigorous result for the PMF - we are more interested in
qualitative results. I've never used the RMSD as a collective variable. I
see there is documentation on how to do this here:
http://www.ks.uiuc.edu/Research/namd/2.9/ug/node55.html#SECTION0001322150000000000000
I also saw that there was some previous discussion on how to do this on the
mailing list:
http://www.ks.uiuc.edu/Research/namd/mailing_list/namd-l/12123.html
The user mentions that he is following the tutorial for ubiquitin. I found
a tutorial here:
http://www.ks.uiuc.edu/Training/CaseStudies/pdfs/ubq.pdfHowever, it
seems that the only colvar that is used is the end-to-end
distance and not the RMSD. Is there another tutorial available?
In the meantime we will try to follow the instructions in the user guide
and perhaps we can get it to work on the first try. I'm just wondering if
there are any other caveats that I need to worry about when using this type
of colvar.
Thanks,
Bob
On Wed, Jun 20, 2012 at 7:25 AM, Jérôme Hénin <jhenin_at_ifr88.cnrs-mrs.fr>wrote:
> Hi Bob,
>
> As you've noticed, the coordinate you used so far gives ambiguous
> results because your system has a lot of flexibility, and will visit
> basins that are not of interest to you. Now there are two kinds of
> approaches to this problem:
>
> 1) add restraints that forbid visiting the unwanted states, but this
> changes the meaning of the PMF you are calculating
> 2) change your set of coordinates to describe the space of interest
> more explicitly, and explore precisely that
>
> In many cases where you want mostly qualitative information on a
> precise process, the first choice is the best one. Trying to extract a
> PMF that is quantitative and meaningful and can yield real free energy
> differences can be very demanding.
>
> Now about finding coordinates that describe the process: one simple
> coordinate that would discriminate between the states that you mention
> is the RMSD of the whole dimer with respect to the hybridized state.
> Since the adsorbed state seems to be a deep and broad well, it doesn't
> seem to need a very precise description to be visited in the
> simulation.
>
> Caveat: finding good coordinates is difficult for us, because we don't
> have the degree of physical intuition that you have about this system,
> its degrees of freedom, and what type of motion is relevant or
> irrelevant to your problem.
>
> Cheers,
> Jerome
>
> On 19 June 2012 22:43, Robert Johnson <robertjo_at_physics.upenn.edu> wrote:
> > Hello All,
> >
> > I'm interested in determining how two complementary DNA strands can
> > hybridize when they are both adsorbed to a carbon nanotube.
> >
> > I have already performed some ABF calculations to estimate the PMF for
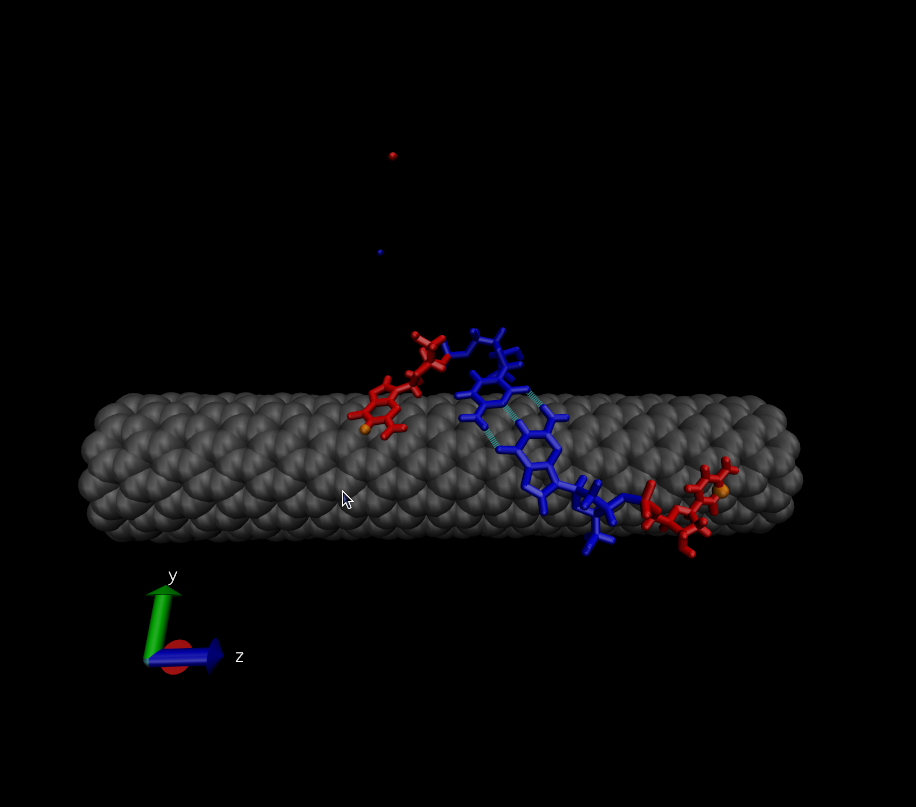
> > hybridization. My initial state is shown here:
> > http://www.physics.upenn.edu/~robertjo/temp/InitialState.png
> >
> > My system consists of 2 DNA strands that are each 2 bases long - in this
> > case each strand is GC. The blue bases are forming a G-C base pair. Over
> the
> > course of the simulation I constrain the distances between the H-bond
> donors
> > and acceptors for this base pair. Therefore, the blue base pair is
> present
> > throughout the entire simulation.
> >
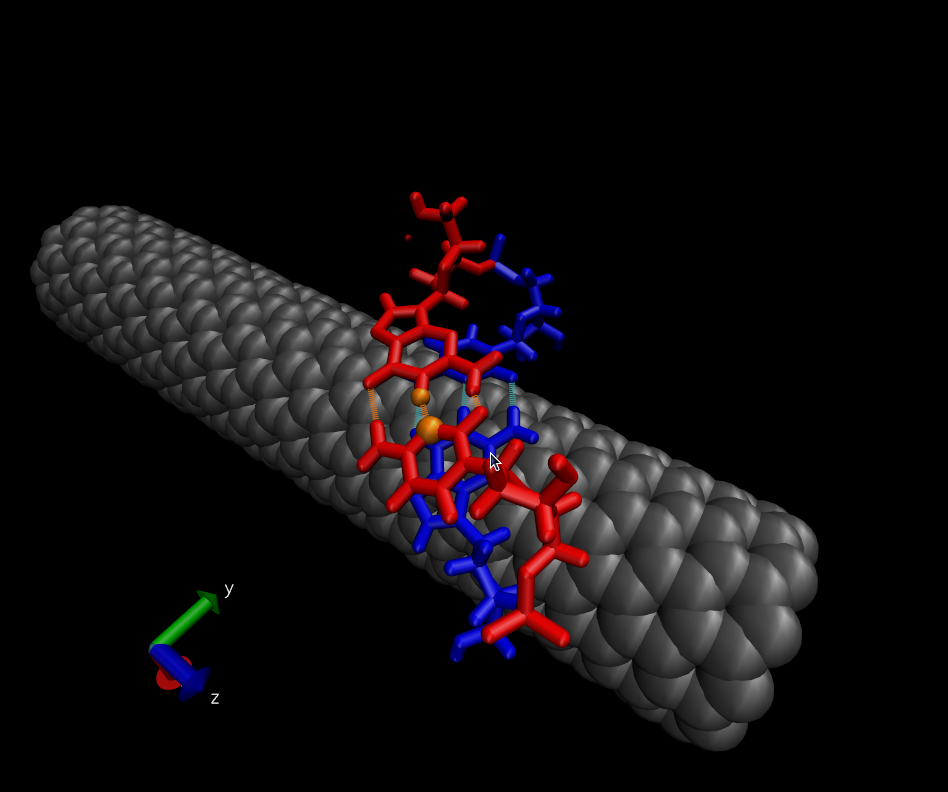
> > Then ABF is employed to force the two red bases to come together. The
> > collective variable used is the distance between two atoms that share a
> > H-bond when the red bases are paired (the orange atoms). Applying ABF
> causes
> > (in most cases) the red bases to move toward each other and to form a
> base
> > pair. The only way the red bases can hybridize is by lifting off the
> surface
> > of the nanotube. The final state is is shown here:
> > http://www.physics.upenn.edu/~robertjo/temp/Hybridized.png
> >
> > A graph of a representative PMF of this process is shown here:
> > http://www.physics.upenn.edu/~robertjo/temp/RepresentativePMF.jpg
> >
> > The 2 strands initially start off in a deep energy minimum corresponding
> to
> > adsorption to the nanotube. Forcing the two red bases to hybridize
> requires
> > the system to surmount a large energy barrier. Then the system falls
> into a
> > small energy minimum as the bases hybridize.
> >
> > About 60% of the time, I obtain a similar structure (and PMF) to that
> shown
> > in the image(s). However, the rest of the time the bases come together
> in an
> > orientation that does not favor hybridization. This makes it a little bit
> > difficult to analyze the results since it is not known ahead of time what
> > pathway the molecules will take.
> >
> > DNA is very flexible and I doubt that I will be able to fully sample all
> the
> > different pathways that the DNA takes to reach the hybridized state.
> > However, I would like a more reliable method for forcing the system to
> reach
> > this hybridized state.
> >
> > Does anyone have ideas for better collective variables to use? Would a
> > different method (i.e. metadynamics or steered MD) be a better choice?
> Since
> > I'm interested in a very specific final state, I've also considered
> starting
> > the simulation from the hybridized state and forcing the strands apart.
> >
> > I would appreciate any feedback you could give. Thanks!
> > Bob
> >
> > --
> > Bob Johnson, PhD
> > Lab Coordinator & Lecturer
> > Department of Physics and Astronomy
> > University of Pennsylvania
> > 209 S. 33rd St.
> > Philadelphia, PA 19104
> > Office: David Rittenhouse Laboratory 2C11
> > Phone: 215-898-5111
> > http://www.physics.upenn.edu/~robertjo
>
-- Bob Johnson, PhD Lab Coordinator & Lecturer Department of Physics and Astronomy University of Pennsylvania 209 S. 33rd St. Philadelphia, PA 19104 Office: David Rittenhouse Laboratory 2C11 Phone: 215-898-5111 http://www.physics.upenn.edu/~robertjo
This archive was generated by hypermail 2.1.6 : Mon Dec 31 2012 - 23:21:41 CST
{kind=link}
{kind=link}
{kind=link}