

Next: Parameter generation using SPARTAN
Up: Parameterization Tutorial
Previous: The CHARMM Force Field
Subsections
When presented with a nonstandard group, the first
task is to determine exactly which parameters are already known,
and which will need to be developed from scratch. The unparameterized part of
the CYG residue is the connection between the sulfur and the carbon. The
connection between the sulfur of CYS and the carbonyl carbon of the glutamine is called a
thioester bond. By attaching the substrate in such a way, you have created a glutamyl
thioester intermediate. In order to use this CYG residue in MD simulations, we need to make a topology
file entry for it as well as develop a complete set of parameters describing it.
To do this, we will integrate two pre-existing topology/parameter file entries -
a cysteine entry and glutamine entry. From a chemical and biological standpoint, this should make
intuitive sense: though connected in a new way, we can still expect the two bonded residues to behave
(for the most part) as they would when they are separate entities. The chemical and physical behavior
of CYG should be similar to CYS and GLN.
The topology file specifies the partial charge of each atom and the connectivity
of the atoms. We will then use the topology file when we create the .psf and .pdb file for our
simulations. The parameter file specifies the specific equilibrium bond lengths and angles, bond and angle
force constants, dihedrals, and impropers. We need bond, angle, and dihedral parameters for
the atoms around this new residue, and we will generate them in the next exercise. The creation of
both the topology file entry and the missing parameters are discussed in detail now.
Below is the final topology file entry for the CYG residue. The CYG parameters
appearing in it were created by combining a
regular CYS entry (with the HG1 atom of CYS deleted) with a GLN entry
(with the NH2 atom of GLN deleted).
When merging the two residues into one, at the place where you connect
the groups you will have to delete atoms (in this case a hydrogen on cysteine's S and the NH2
atom of the glutamine)
and alter the charges. Following the standard CHARMM protocol, the HG1 atom
on CYS was deleted and its charge was moved to SG (-0.23 + 0.16 = -0.07).
In the CYG entry, you can see the line for HG1 has been commented out (comments
are denoted by ! marks), and the charge added to the S. Note that the group
still has an overall neutral value.
Take a few minutes and look through one of the topology files we have supplied you.
To do this, at a unix prompt, type:
more top_all27_prot_lipid_prfar_cyg_cyn.inp
Look at a
regular CYS and GLN residue entries, and be sure you understand how we have pasted them
together to make CYG below.
RESI CYG 0.00
GROUP
ATOM N NH1 -0.47 ! |
ATOM HN H 0.31 ! HN-N
ATOM CA CT1 0.07 ! | HB1
ATOM HA HB 0.09 ! | |
GROUP ! HA-CA--CB--SG
ATOM CB CT2 -0.11 ! | | |
ATOM HB1 HA 0.09 ! | HB2 |
ATOM HB2 HA 0.09 ! O=C |
ATOM SG S -0.07 ! | \
!ATOM HG1 HS 0.16 ! \
GROUP ! \
ATOM CDG CC 0.55 ! \
ATOM OE1 O -0.55 ! \
GROUP ! HN2G \
ATOM CGG CT2 -0.18 ! | \
ATOM HG1G HA 0.09 ! HN1G-NG HB1G HG1G\
ATOM HG2G HA 0.09 ! | | | \
GROUP ! HAG-CAG--CBG--CGG--CDG=OE1
ATOM CBG CT2 -0.18 ! | | |
ATOM HB1G HA 0.09 ! | HB2G HG2G
ATOM HB2G HA 0.09 ! O1G=CG
GROUP ! |
ATOM CG CD 0.75 ! O2G-HO2G
ATOM O1G OB -0.55
ATOM O2G OH1 -0.61
ATOM HO2G H 0.44
ATOM CAG CT1 -0.12
ATOM HAG HB 0.09
ATOM NG NH3 -0.62
ATOM HN1G HC 0.31
ATOM HN2G HC 0.31
GROUP
ATOM C C 0.51
ATOM O O -0.51
BOND CB CA SG CB N HN N CA
BOND C CA C +N CA HA CB HB1
BOND CB HB2
BOND SG CDG CDG CGG CGG HG1G CGG HG2G
BOND CGG CBG CBG HB1G CBG HB2G
BOND CBG CAG CAG HAG CAG NG CAG CG
BOND NG HN1G NG HN2G CG O2G O2G HO2G
DOUBLE O C CDG OE1 CG O1G
IMPR N -C CA HN C CA +N O
IMPR CDG OE1 SG CGG CG O1G O2G CAG
DONOR HN N
DONOR HN2G NG HN1G NG HO2G O2G
ACCEPTOR O C OE1G CDG O1G CG O2G CG NG CAG
Note the following features in the CHARMM27 topology file:
- In the second column you can find the atom name of each individual atom. The connectivity of
the atoms is depicted in the drawing and uses the atom name. This name must match the atom name
in the pdb file.
- In the third column the atom type is designated for each atom. The parameter files of force fields
do not have chemical properties for individual atoms of the system. Instead, they have parameters for atom types
in order to minimize the number of entries in the file. It is assumed that the chemical properties of similar atoms are
the same, i.e. every carbonyl group of the backbone has similar chemical and physical properties. From this column
the force field extracts the information for assigning the right parameters during the simulation. If you want to
distinguish two chemically functional groups from each other, then you have to give them different atom types in the
force field. Assigning only different atom names is not sufficient.
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2...
... and torsional terms be the same
for both functional groups? }
\end{minipage} }](img29.gif)
- The fourth column shows the partial charge of each atom. There are various quantum mechanical methods
available for assigning partial charges in force fields. We will discuss the determination of partial charges in
more detail later. The atoms are divided into groups; with the exception of charged residues (such as arginine or glutamate),
the net charge of each group is neutral.
- At the end of the entry, you will find Bond (BOND)and Improper (IMPR) entries.
These entries define the connectivity and the planarity of residues.
You may have noticed that although the bonds and atoms are defined in the topology file,
the entry is missing angle and dihedral assignments. The angle and dihedrals are never assigned in the topology file.
PSFGEN automatically determines the angles and dihedrals based on the connectivity of the atoms. This should
make sense - every 3 connected atoms generates an angle and every 4 generates a dihedral term. By automatically
generating the terms, PSFGEN saves you a lot of work!
Below is an excerpt from a CHARMM27 parameter file that
shows some required (and previously missing!) parameters necessary for MD
simulations.
The force field excerpts below contain the final missing heavy atom
parameters for CYG and can be used as a reference for the
assessment of the parameters you develop in the next few
exercises. Due to time constraints, for the following exercises,
we will only perform semi-empirical calculations. The final
parameter set was developed using a higher level of theory (an ab
initio Hartree-Fock calculation with the 6-31G* basis set).
Therefore the final parameter values presented below may differ
slightly than the ones you determine.
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2...
... pentane
(both systems have the same number of heavy atoms)? }
\end{minipage} }](img32.gif)
BONDS
!
!V(bond) = Kb(b - b0)**2
!
!Kb: kcal/mole/A**2
!b0: A
!
!atom type Kb b0
! Modified for CYG residue after 6-31G* geometry optimization
S CC 240.000 1.7814 ! ALLOW ALI SUL ION
ANGLES
!
!V(angle) = Ktheta(Theta - Theta0)**2
!
!V(Urey-Bradley) = Kub(S - S0)**2
!
!Ktheta: kcal/mole/rad**2
!Theta0: degrees
!Kub: kcal/mole/A**2 (Urey-Bradley)
!S0: A
!
!atom types Ktheta Theta0 Kub S0
!
! Modified for CYG residue after 6-31G* geometry optimization
CT2 S CC 34.000 100.2000 ! ALLOW ALI SUL ION
CT2 CC S 50.000 114.5000 ! ALLOW ALI SUL ION
O CC S 75.000 122.2000 ! ALLOW ALI SUL ION
DIHEDRALS
!
!V(dihedral) = Kchi(1 + cos(n(chi) - delta))
!
!Kchi: kcal/mole
!n: multiplicity
!delta: degrees
!
!atom types Kchi n delta
CC S CT2 CT1 0.2400 1 180.00
CC S CT2 CT1 0.3700 3 0.00
HA CT2 S CC 0.2800 3 0.00
CT2 S CC CT2 2.05 2 180.00
CT2 S CC O 2.05 2 180.00
Note the following features in the CHARMM27 parameter file:
- It contains all numerical values required for CHARMM27 energy functions
explained in section 2 of this tutorial. The force field contains
entries for bonds, angles, dihedrals, impropers and nonbonded
interactions. The force field also contains comments describing
where these values have been obtained either by experimental
spectroscopic measurements or quantum mechanical calculations.
- The bond and angle entries contain the value for the force constant
in the first column in the physical units of kcal/mole/Å. The
second column contains equilibrium bond or angle distance in Å
or degrees, respectively.
- The next section contains the torsion (a.k.a. dihedral) angles. The individual values
will be explained in the course of the exercise and you will perform a dihedral drive of the
dihedral HA CT2 S CC.
The values for the nonbonded interactions are the standard
VDW parameters in CHARMM27 required for the Lennard-Jones
potential of your atom types.
Before we get into the details of parameterizing, it is useful and often very
helpful to be aware of programs that are able to assist you in the process.
CHARMm, AMBER, and MOE are all programs with modules that
will identify which atoms are missing parameters and even give you an initial
first guess of values. These programs almost always use a pattern recognition
algorithm to guess for you, and any parameters they supply you will have to be
checked with your chemical intuition and at least a basic level of quantum mechanics.
If you choose to use one of these packaged programs, caveat emptor! While
it seems like an easy solution to a sometimes difficult and always tedious process,
it rarely, if ever, provides you with a suitable and/or reliable set of parameters.
Note the dihedral (torsional) parameters. The analytical form of a dihedral term in the CHARMM force field
uses cosine functions:
![\begin{displaymath}
V_\phi = k_\phi \left[ 1+ \left( \cos \left(n\phi-\delta\right)\right)\right]
\end{displaymath}](img33.gif) |
(2) |
where 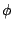 is the value of the dihedral angle,
is the value of the dihedral angle, 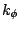 is the
force constant, n is the symmetry of the rotor (e.g. 3 for methyl
groups) and
is the
force constant, n is the symmetry of the rotor (e.g. 3 for methyl
groups) and 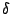 is the phase. In the CHARMM energy function,
you can specify multiple dihedrals for the same bond. Often one needs to
use multiple dihedrals in order to generate energetic barriers of proper height
and location.
is the phase. In the CHARMM energy function,
you can specify multiple dihedrals for the same bond. Often one needs to
use multiple dihedrals in order to generate energetic barriers of proper height
and location.
You can use the following table to help you generate the correct dihedrals in CHARMM.
| Dihedral Parameters in CHARMM format |
| Multiplicity, n |
Phase |
Location of Minima |
Useful Notes |
| 1 |
0 |
180 |
yields trans conformation |
| 1 |
180 |
0 |
yields cis conformation |
| 2 |
0 |
90, 270 |
|
| 2 |
180 |
0, 180 |
useful for enforcing planarity |
| 3 |
0 |
60, 180, 300 |
emphasizes staggered conformation of sp3 carbons |
| 3 |
180 |
0, 120, 240 |
emphasizes eclipsed conformations of sp3 carbons |
For instance, the line:
HA CT2 CT2 CT3 3 0.195 0
could also be written, for these four atoms:
![\begin{displaymath}
V_\phi = 0.195\cdot\left[1+\cos\left(3\phi\right)\right]
\end{displaymath}](img34.gif) |
(3) |
i.e., the four atom types, the symmetry of the rotor (n), the force constant () and the phase (d).
Since we only have a force constant for n=3, the value of the
force constant is 0.195, and the phase is zero. A sample Matlab plot is shown in Fig. 4.
Figure 4:
Matlab plot of Eq. (3) presented
above.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[scale=0.5]{FIGS/dihedral}
}
\end{center}
\end{figure}](img35.gif) |
Next: Parameter generation using SPARTAN
Up: Parameterization Tutorial
Previous: The CHARMM Force Field
tutorial@ks.uiuc.edu
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2...
...ostly neutral? How is this advantageous for an MD simulation?}
\end{minipage} }](img30.gif)
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2...
... Which line(s) specify the new bonds present in this residue?}
\end{minipage} }](img31.gif)