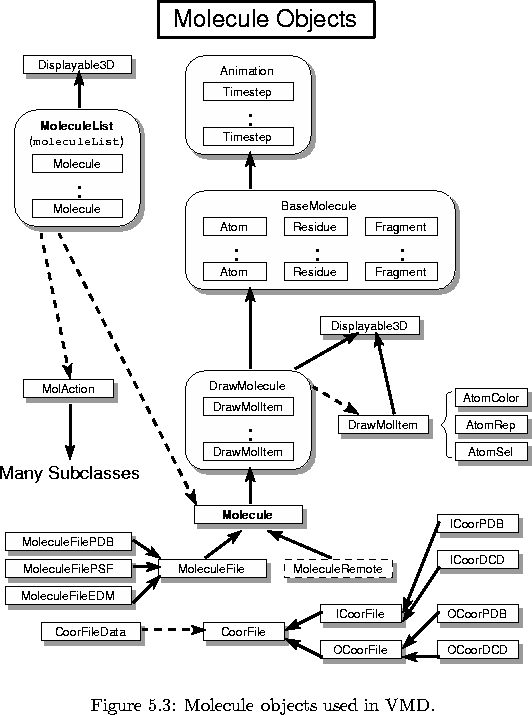
The objects used for displaying and rendering graphical objects in VMD are quite general, and can be used to draw essentially anything. The objects used by VMD to create, store, and manipulate molecules, which are illustrated in figure 5.3, are much more specific to the purpose of VMD, which is to visualize to dynamic properties of biopolymers (in particular proteins and nucleic acids). The heart of this category of object classes is the Molecule class, which is actually inherited from a number of base classes and for which several subclasses exist. MoleculeList is an object which maintains a list of all current molecules. There are also several helping objects which store data about particular components of each molecule.
At the very top level of the Molecule hierarchy is the Animate class, which stores a list of Timestep objects. Nothing is known about the molecule at this level other than the number of atoms; a Timestep stores simply arrays of floating-point values for each of these atoms for each discrete timestep in the trajectory of the molecule. The Animate class also maintains the current frame in the trajectory, and the direction (i.e., fast-forward, reverse, pause) and speed of animation. The Timestep objects, one for each frame of animation in each molecule, are stored simply as pointers in a ResizeArray instance within the Animate object. A Timestep is currently quite simple, and stores data as publicly-available floating point arrays which are allocated when a new Timestep is created (or for some data which may be optionally stored for each step, by the users request). This is done primarily for speed since this data is accessed quite often. It may be helpful to improve this class in the future, by making a much more general method to store different types of atomic data for timesteps which would not require a change to the Timestep class each time. For example, the Timestep may just store a list of pointers to something like a TimeStepData class instance, where each TimeStepData would store some number of floating-point values.
At the next level, the BaseMolecule object is inherited from the Animate object. This object stores all the basic information which comprises the structure of the molecule. Data about the coordinates are stored by Animate, while BaseMolecule stores how the atoms are connected, what residues and segments exist, etc. When it is created, a BaseMolecule is empty, indicating no atoms or anything present. A virtual function BaseMolecule::() is used by BaseMolecule and all other class derived from BaseMolecule; this function is called when a new molecule is to be created, and derived classes do their creation tasks after which they call create for the parent class. BaseMolecule does NOT contain any data or functions for the drawing of the molecule, just for storing the structure. After a new molecule has been read in from some files or from a network connection, the structure of the molecule is analyzed and stored in a retrievable format. Several small classes help in this storage; they include the following:
From BaseMolecule and from Displayable3D the DrawMolecule object is derived. This level of the Molecule hierarchy stores all the information about how to draw the molecule. A molecule in VMD is drawn as a composition of one or more representations of the molecule structure, which are contained within a DrawMolItem object (described later). A DrawMolecule stores a list of all the different representations (DrawMolItem) of the molecule that the user has selected, and contains routines to add, change, or delete these representations. Since it is also a Displayable, a DrawMolecule can specify its own display list, but currently all molecule drawing commands are contained within DrawMolItem objects.
A DrawMolItem is also derived from Displayable3D; its function is to maintain the display list with the proper drawing commands to render one specified representation of a molecule. When a new DrawMolItem is created, it is given the molecule for which it is to render an image, and instances of the following three objects which describe exactly what the representation is to be:
The main base class Molecule is then derived from DrawMolecule. This is the level at which most other objects in VMD work with molecules, as pointers to instances of a Molecule class. In fact relatively little functionality is includes at this level. What this class does do, in fact, is provide the routines for reading in or writing out of animation frames from or to different coordinate file formats (i.e., PDB or DCD files). This is done through the use of a CoorFileData object, which encapsulates the information on how to read/write such a coordinate file (this includes storing which frames are to be read or written, the coordinate file format, and the current status of such an operation). Since Molecule is a subclass of Displayable, it has a prepare virtual routine which is called each time the Scene is to be drawn to the current DisplayDevice. Molecule uses this call to prepare to read/write a single coordinate set from/to the current coordinate file, if one is being processed. Thus, a coordinate file is not processed in one single operation, instead one frame is processed each time the Scene is drawn. This allows VMD to continue to animate and check for user commands while a coordinate file is being read or written. For the actual coordinate file reading or writing, the CoorFile base class and derived ICoorFile and OCoorFile classes abstract the action of taking a set of XYZ positions for a molecule and reading or writing a trajectory file. Specific versions of these classes for PDB and DCD files are used, and any other number of trajectory formats may be supported by developing new subclasses of ICoorFile and OCoorFile, with also an update to CoorFileData.
Up to the point just described are all the classes necessary to store and manipulate a molecule. However, there are several different ways for a molecule to be imported into VMD, and each method has a specific subclass of Molecule to provide the functions to read in the proper data and store it into the standard internal format of the Molecule class hierarchy. Currently, the following subclasses of Molecule exist:
Thus, the steps in creating a new molecule in VMD are as follows:
The object which keeps track of all the currently-loaded Molecule objects is MoleculeList, of which there is exactly one in VMD assigned to the global variable moleculeList (although there is no reason why there could not be more than one). MoleculeList is an important object: it manages all the molecules, contains routines to allow an operation to be performed on a number of molecules at the same time, and supplies information on how the molecules are related to each other. MoleculeList is derived from Displayable as well: it is the top-level parent Displayable with all Molecule objects as child Displayables. Thus, turning off the MoleculeList turns off all the molecules, and similarly rotating or scaling the MoleculeList does so to all the molecules. There are no drawing commands currently for the MoleculeList itself (although they could be added for something which indicates relationships between the molecules). This is a useful trick in VMD: have a container class which is derived from Displayable, but which has no drawing commands of its own; instead, have it contain several child Displayable objects which form components of the complex object which is to be drawn. By applying rotations, translations, etc. to the container class, all the child components are similarly transformed, and they may be separately altered or turned on or off.