The plugin helps you to generate the molecule or the moleular fragment that should be parametrized and to set up the necessary quantumchemical calculations (you'll need Gaussian, later versions will also support GAMESS). Paratool reads the logfiles of the QM simulations, computes force field parameters for bonds, angles dihedrals and impropers by transforming the Hessian into internal coordinates and converting all values into units based on kcal/mol, Å and degrees. You can assign atom types and the corresponding VDW parameters from a list of already existing types or generates new types (all chemical elements H - Rn supported). The charges can be determined using restricted electrostatic potential fitting RESP (AMBER style) or CHARMM style charges using their supramolecular approach which requires another Gaussian calculation.
Paratool generates input for all necessary external programs collects and compiles all the necessary data, organizes and displays them neatly in lists and projects them onto your molecule in VMD. Finally it will write the topology and parameter files you'll need to build the molecule using psfgen and run your simulation in your favourite simulation package, e.g. NAMD.
Parametrizing a novel component requires the following steps which will be explained in detail further below: First a molecular model compound containing unparametrized parts has to be defined. Then the atom type and its according VDW parameters have to be chosen for each atom from a list of existing atom types of the selected force field. Further, all transferable parameters from the existing force field have to be assigned to the model. If missing parameters remain then these will be determined de novo. In that case the model compound is the starting point for a quantumchemical geometry optimization. A QM frequency calculation based on the optimized geometry yields the Hessian matrix which can be transformed into the set of internal coordinates describing the bonded interactions. The diagonal elements of this internal Hessian denote the force constants related to these coordinates. The basis for the charges is obtained from a QM single point calculation from which Mulliken charges, natural population analysis (NPA) charges [7] and an electrostatic potential (ESP) charge fit [1,8] can be extracted. These raw data can subsequently be used to compute the CHARMM type charges according to the supramolecular approach or to setup a restricted electrostatic potential fit (RESP). Subsequently, the parameters should be refined in order to account for the implicit double accounting of electrostatics in 1-4 interactions. Finally the new parameters must be tested in a molecular dynamics simulation.
The current version of Paratool only considers quantum chemical target data such as equilibrium geometry, vibrational frequencies, electrostatic field and dipole moment. Nevertheless, you can manually replace the quantum chemical equilibrium geometries and force constants for bonds, angles, dihedrals and impropers by experimental values.
Paratool does not yet implement the aforementioned automatic selfconsistent iterative fitting against these data but you can of course manually loop through several cycles of refinement and hand-tune the data any time. Though in many cases the first iteration will already yield satisfactory results.
top_all27_prot_lipid_na.inp and
par_all27_prot_lipid_na.inp containing amino acids,
nucleic acids, lipids and a few other common compounds. But there's
more to find in the CHARMM files. Especially take a look at the file
for the model compounds top_all22_model.inp.
RESI ASP -1.00 GROUP ATOM N NH1 -0.47 ! | ATOM HN H 0.31 ! HN-N ATOM CA CT1 0.07 ! | HB1 OD1 ATOM HA HB 0.09 ! | | // GROUP ! HA-CA--CB--CG ATOM CB CT2 -0.28 ! | | \ ATOM HB1 HA 0.09 ! | HB2 OD2(-) ATOM HB2 HA 0.09 ! O=C ATOM CG CC 0.62 ! | ATOM OD1 OC -0.76 ATOM OD2 OC -0.76 GROUP ATOM C C 0.51 ATOM O O -0.51It is stitched together from two smaller model compounds acetate and glycine (found in
top_all22_model.inp and top_all27_prot_lipid.inp) where H2 and HA2 are removed and C1 and CA are connected by a bond (C1 then become CB in the new compound etc.)
The charges of the two removed hydrogens were simply added to their mother atoms.
RESI ACET -1.00 ! acetate, K. Kuczera GROUP ATOM C1 CT3 -0.37 ! ATOM C2 CC 0.62 ! H1 O1 (-) ATOM H1 HA 0.09 ! | / ATOM H2 HA 0.09 ! H2--C1--C2 ATOM H3 HA 0.09 ! | \\ ATOM O1 OC -0.76 ! H3 O2 ATOM O2 OC -0.76 !
RESI GLY 0.00
GROUP
ATOM N NH1 -0.47 ! |
ATOM HN H 0.31 ! N-H
ATOM CA CT2 -0.02 ! |
ATOM HA1 HB 0.09 ! |
ATOM HA2 HB 0.09 ! HA1-CA-HA2
GROUP ! |
ATOM C C 0.51 ! |
ATOM O O -0.51 ! C=O
! |
But where do the parameters for the peptide bonds that are formed when
the multiple residues are joined in a polypeptide come from?
They were obtained from the model compound N-methylacetamide. For CR
the peptide bond would be in N-terminal position for CL it would be C-terminal.
RESI NMA 0.00 ! N-methylacetamide, Louis Kuchnir
GROUP
ATOM CL CT3 -0.27 ! HR2
ATOM HL1 HA 0.09 ! |
ATOM HL2 HA 0.09 ! HR1--CR--HR3
ATOM HL3 HA 0.09 ! |
ATOM C C 0.51 ! |
ATOM O O -0.51 ! N-H
ATOM N NH1 -0.47 ! |
ATOM H H 0.31 ! O=C
ATOM CR CT3 -0.11 ! |
ATOM HR1 HA 0.09 ! |
ATOM HR2 HA 0.09 ! HL1--CL--HL3
ATOM HR3 HA 0.09 ! |
! HL2
If one combines two NMA molecules the in the same manner we merged GLY
and ACET we obtain a model compound with two peptice bonds. Clipping out the middle amino acid at the
peptide bonds yields GLY as seen above.
Try to split your molecule into fragments that are parametrized
already! This saves a lot of time, you might end up with a much
smaller and simpler molecule to parametrize which you then combine
with the other pieces. This is also makes your parameters more
CHARMM compliant. Unfortunately Paratool does not yet support
automatic combining of model compounds so you'll have to do that last
step manually.
One thing has to be kept in mind when selecting the model
compounds. When they are combined then parameters spanning the linkage
like the link bond and the involved angles have to be present,
too. However in most cases these links are something simple like a
bond between two unpolar sp3 hybridized carbons for which numerous
templates exist.
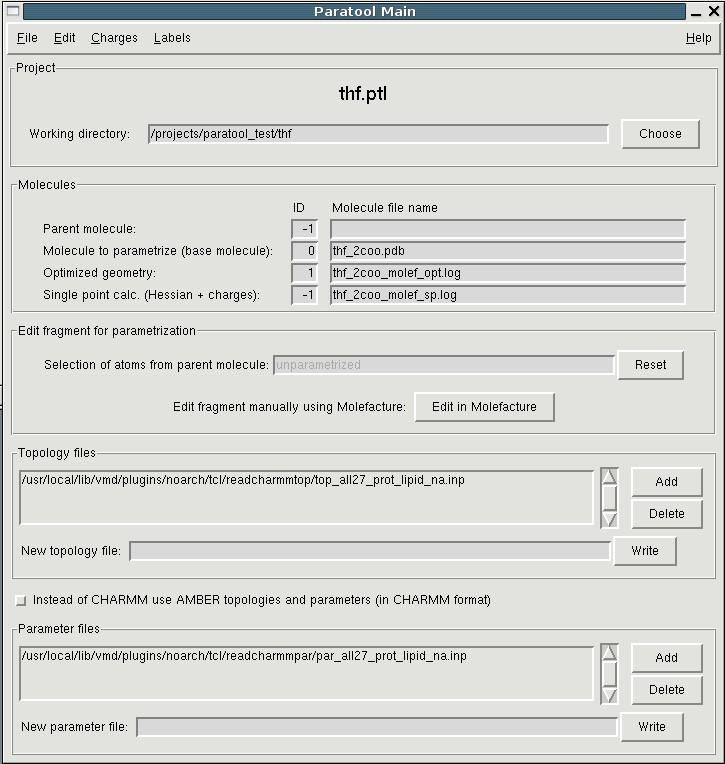
To begin a new Paratool project you can simply start Paratool from the
Tkcon command line or from the Extensions Menu in VMD's Main window.
In the GUI that pops up (see fig. 1), you'll see entries for IDs and file names for
4 different molecules. The ID is the same as in the VMD Main window.
The molecular model compound to which all further work will be related
and into which all data will be displayed is the "base molecule". You
can load it using the File menu. In the file open dialog you can
choose between different file types. PDB is the default. XBGF is an
internal file format derived from "biograf". It is similar to the PDB
format, but is able to store additional info such as atom types, VDW
parameters and partial charges.
Throughout the parametrization process a quantum mechanically (QM) optimized structure of the base molecule and a QM single point calculation will be generated. They'll populate the last two entries.
The parent molecule is the molecule embedding the base molecule, e.g. the protein containing a nonstandard aminoacid or an organic substrate. The parent molecule is actually only needed if there are covalent bonds or coordination complex bonds (e.g. metal complexes or iron-sulfur clusters) exist between the unparametrized part and the rest of the molecule. In this case the size of the environment which is added to the base molecule can be controlled interactively using the entry labelled with "Selection of atoms from parent molecule:" in the Paratool Main window. The parent molecule is set automatically if you start Paratool using AutoPSF.
While it is probably a good idea to prepare the model compound beforehannd in your favourite molecule editor you can also edit your base molecule on the fly using VMD's built in editor Molefacture. You can for example cap open valences where covalent bonds were chopped or delete atom groups that you want to parametrize separately. I'll repeat myself here: Generally you should try to split larger molecules into smaller fragments and parametrize these separately. You can save a lot of CPU time during the quantumchemical calculations and if the fragments are chosen properly you can generate transferable parameters for molecular building blocks. The CHARMM and AMBER force fields are both designed for this additive approach. These model compounds can finally be combined to real molecules again.
Note that whenever you change the selection of the base molecule or you modify it using Molefacture the previous base molecule will be deleted and a new one generated and loaded. Thus the molecule ID will not stay the same.
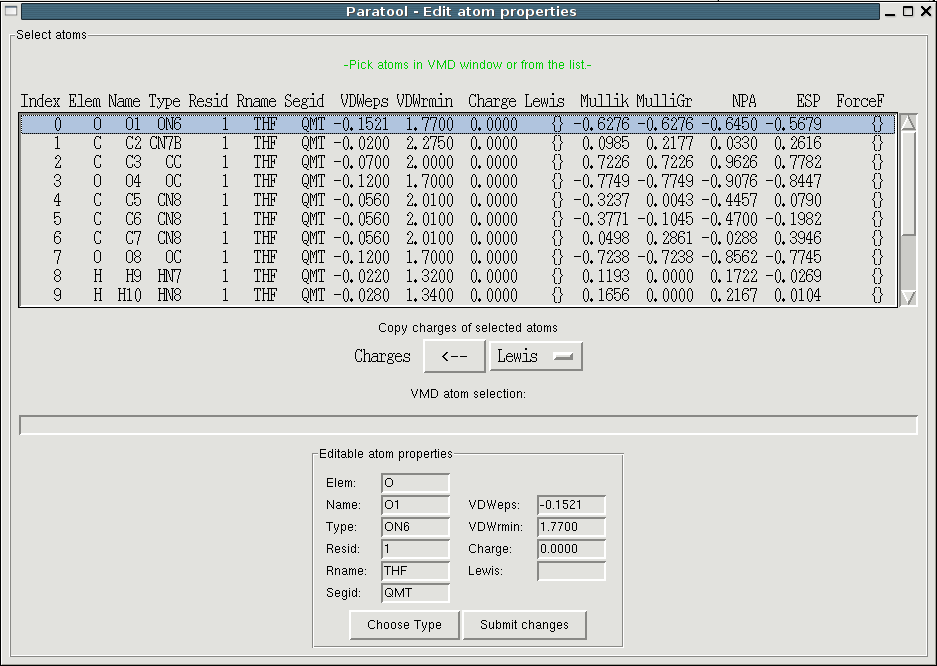 Now that your model compound has been determined, you can already set
many atom based properties. Use "Edit->Atom properties" to open the
appropriate interface. You'll see a list of properties for each atom
from which you can select single or multiple atoms to edit them using
the entry fields at the bottom of the window. There are three ways of
selecting atoms. 1) You can directly click on atoms in the OpenGL
window, the atom becomes marked with an orange sphere and seleccted in
the listbox. Holding the shift key while picking adds the atom to the
selection rather that replacing it. 2) You can select atoms from the
list using the mouse or the arrow keys. Dragging or shift selects
contiguous groups from the list, holding the control key allows to
select arbitrary atoms. 3) You can use VMD's atom selection language.
Now that your model compound has been determined, you can already set
many atom based properties. Use "Edit->Atom properties" to open the
appropriate interface. You'll see a list of properties for each atom
from which you can select single or multiple atoms to edit them using
the entry fields at the bottom of the window. There are three ways of
selecting atoms. 1) You can directly click on atoms in the OpenGL
window, the atom becomes marked with an orange sphere and seleccted in
the listbox. Holding the shift key while picking adds the atom to the
selection rather that replacing it. 2) You can select atoms from the
list using the mouse or the arrow keys. Dragging or shift selects
contiguous groups from the list, holding the control key allows to
select arbitrary atoms. 3) You can use VMD's atom selection language.
The first property you want to set is the residue name (Rname). Select all atoms and type a name in the adequate field, confirming with "Submit". Make sure that the residue name is unique among the topology files you are planning to use. The residue number (Resid) and the segment ID (Segid) are only relevant for transition metal complexes and iron-sulfur clusters but they must be identical for all atoms of a model compound. If you are unhappy with the default atom names from you can change them but each name must be unique in a residue. Also note that atom names and the aforementioned strings should be in capital letters.
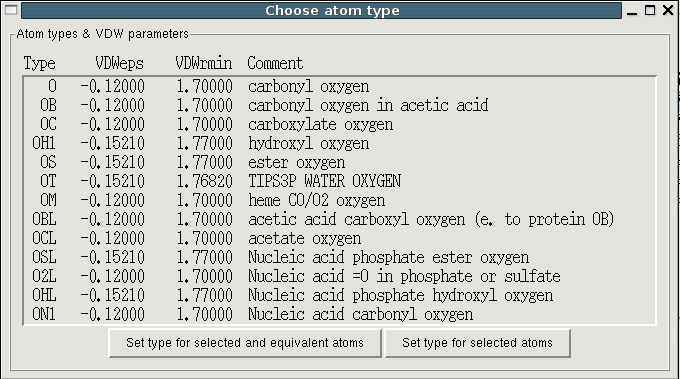 Next you have to choose the atom types. They describe a combination of
periodic element and chemical environment. Thus equivalent atoms such
as hydrogens in methyl groups of carboxylate oxygen have the same
type. In simulations the parameters for all bonded and VDW
interactions are looked up by the atom types.
The user can choose types and VDW parameters from a list of atom types
of the same chemical element existing in the current force field,
i.e. that occur in the loaded parameter files. You'll get there
pressing the "Choose type" button in the "Edit atom properties"
window. It is up to you to decide which given atom type matches the
chemical properties of the selected atom best. If you want these VDW
params but need a different type, you can simply rename it. For
elements that are not contained in the force field there are at the
bottom of the list atom types from the Universal Force Field (UFF)
[6]. This asserts that there are VDW params for
each chemical element from H-Rn. These will also be chosen when you
autoassign VDW parameters from the "Edit" menu.
Here's a simple guideline for the assignments:
Try to find an equivalent existing type of the same chemical element,
Paratool lets you pick that from a list. Many of the types for a certain
element have the same VDW params, e.g. CD and CC.
Next you have to choose the atom types. They describe a combination of
periodic element and chemical environment. Thus equivalent atoms such
as hydrogens in methyl groups of carboxylate oxygen have the same
type. In simulations the parameters for all bonded and VDW
interactions are looked up by the atom types.
The user can choose types and VDW parameters from a list of atom types
of the same chemical element existing in the current force field,
i.e. that occur in the loaded parameter files. You'll get there
pressing the "Choose type" button in the "Edit atom properties"
window. It is up to you to decide which given atom type matches the
chemical properties of the selected atom best. If you want these VDW
params but need a different type, you can simply rename it. For
elements that are not contained in the force field there are at the
bottom of the list atom types from the Universal Force Field (UFF)
[6]. This asserts that there are VDW params for
each chemical element from H-Rn. These will also be chosen when you
autoassign VDW parameters from the "Edit" menu.
Here's a simple guideline for the assignments:
Try to find an equivalent existing type of the same chemical element,
Paratool lets you pick that from a list. Many of the types for a certain
element have the same VDW params, e.g. CD and CC.
C 0.000000 -0.110000 2.000000 ! ALLOW PEP POL ARO
! NMA pure solvent, adm jr., 3/3/93
CA 0.000000 -0.070000 1.992400 ! ALLOW ARO
! benzene (JES)
CC 0.000000 -0.070000 2.000000 ! ALLOW PEP POL ARO
! adm jr. 3/3/92, acetic acid heat of solvation
CD 0.000000 -0.070000 2.000000 ! ALLOW POL
! adm jr. 3/19/92, acetate a.i. and dH of solvation
The reason why there are two types is that they are used in different
contexts so that you can for instance distinguish dihedrals including CC
or CD.
Which type to use has to be decided using chemical insight. Just try to
find the most similar atomtype by comparing structures where it appears.
Factors influencing this similarity are for example hybridization state,
polarization, same neighboring atoms.
Always try to use an existing atom type, unless you end up with bonded
parameters that are not equivalent enough. In the latter case you just
change the type name, but keeping the VDW params. In case there is no type
for the atom's chemical element you can use VDW parameters from the
Universal Force Field (they are at the end of the list). You can of course
change the predefined typename if you want.
Assinging types implies the automatic assignment of bonded parameters for
which all types are known and for which a matching entry could be found in
the currently loaded parameter files.
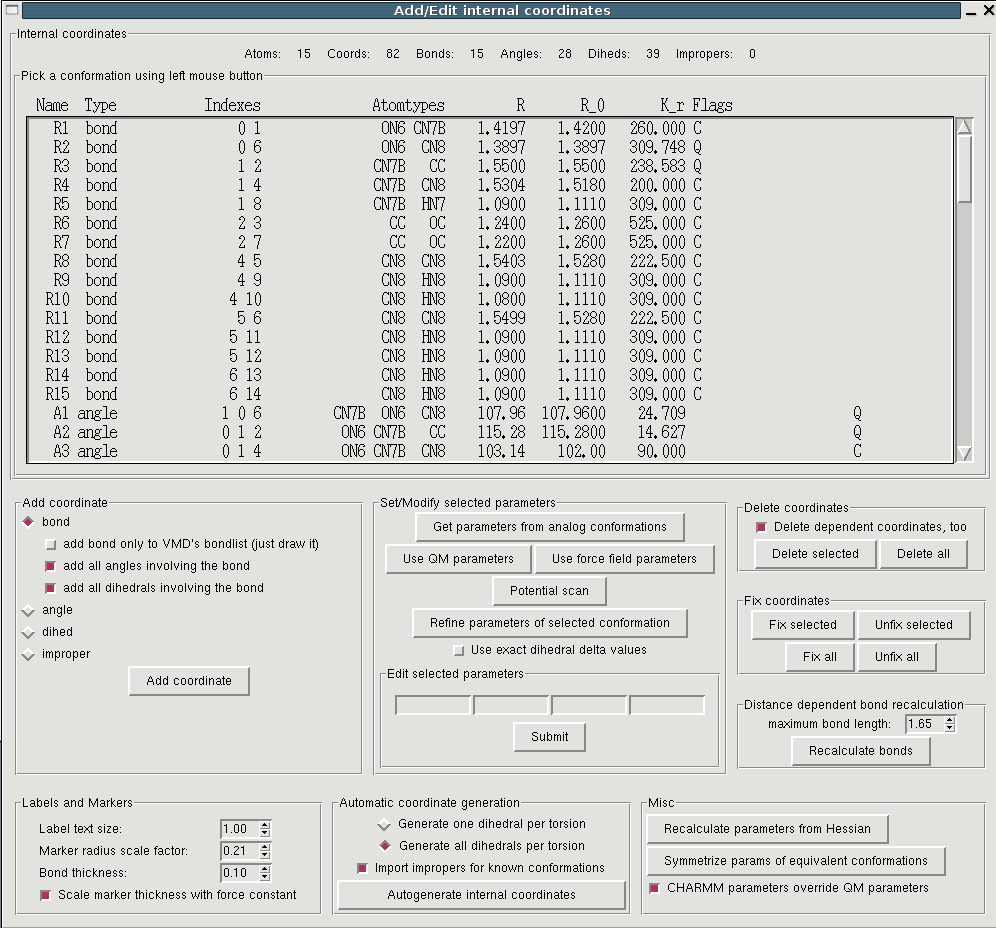 Before bonded parameters can be assigned it has to be specified for
which internal coordinates, i.e. bonds, angles, dihedrals and
impropers bonded interactions must be determined. For each of these
coordinates parameters for bonded interaction will either be taken from
existing model compounds, or, in case no equivalent exists, be derived
from the Hessian matrix (see below). "Edit->Intenal coordinates" opens
a window containing a (initially empty) list of defined internal coordinates
and a plethora of buttons. A good starting point is to autogenerate the
internal coordinates. In this procedure all covalent bonds of a
molecule (as displayed in VMD) are defined as bonds. Then all possible
angles and dihedrals that can be constructed from the given bonds are
generated. In many cases this is already sufficient but you can add
and delete arbitrary coordinates. You can for example add impropers to
stabilize planar geometries. More info about the choice of internal
coordinates can be found here.
Note: After you have generated internal coordinates you can already write
valid topology files using the accordant button in the main window.
Before bonded parameters can be assigned it has to be specified for
which internal coordinates, i.e. bonds, angles, dihedrals and
impropers bonded interactions must be determined. For each of these
coordinates parameters for bonded interaction will either be taken from
existing model compounds, or, in case no equivalent exists, be derived
from the Hessian matrix (see below). "Edit->Intenal coordinates" opens
a window containing a (initially empty) list of defined internal coordinates
and a plethora of buttons. A good starting point is to autogenerate the
internal coordinates. In this procedure all covalent bonds of a
molecule (as displayed in VMD) are defined as bonds. Then all possible
angles and dihedrals that can be constructed from the given bonds are
generated. In many cases this is already sufficient but you can add
and delete arbitrary coordinates. You can for example add impropers to
stabilize planar geometries. More info about the choice of internal
coordinates can be found here.
Note: After you have generated internal coordinates you can already write
valid topology files using the accordant button in the main window.
We can make even more use of the existing force field through parametrization by analogy. In other words we are utilizing parameters of parts that have a similar chemical environment. You must decide what is chemically equivalent using your chemical insight. Among the properties you should compare are periodic elementss of the involved atoms, charges, bond orders and polarity of bonds. This technique has been applied a lot within the force field already. This is the reason why you find so many identical parameter entries. Here's an example:
CT1 CC 200.000 1.5220 ! ALLOW POL CT2 CC 200.000 1.5220 ! ALLOW POL CT3 CC 200.000 1.5220 ! ALLOW POLOnly the type names of the involved atoms differ but the chemical nature of this bond is considered the same. The different type names were needed to distinguish other properties like angles or dihedrals.
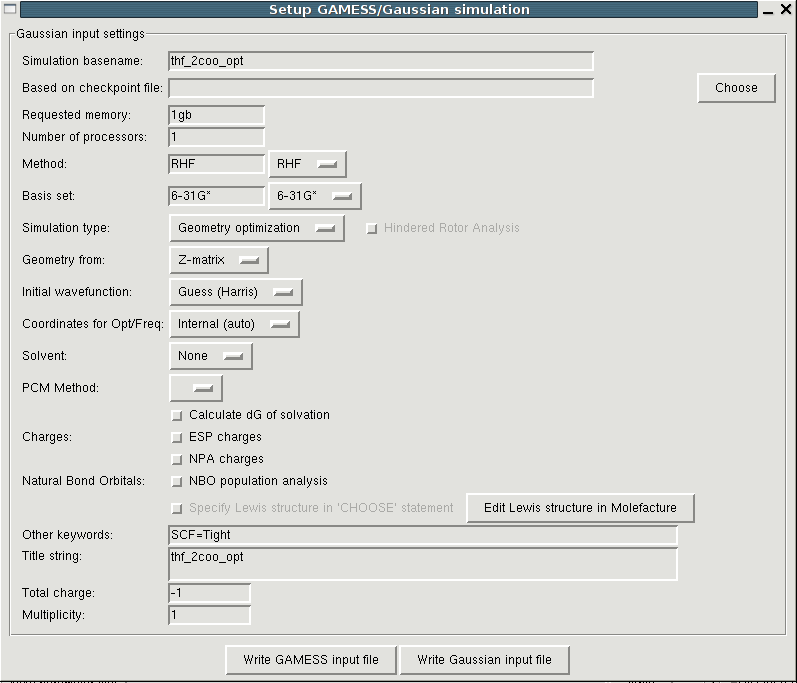 For the calculation of partial charges and the parameters for bonded
interactions we need the QM optimized structure of the base molecule.
In the current version Paratool supports the autogeneration of input
files for Gaussian but later
versions will also provide an interface to
GAMESS.
(For this task Paratool uses
QMtool
which you can also use stand-alone to setup and analyze QM simulations.)
For the calculation of partial charges and the parameters for bonded
interactions we need the QM optimized structure of the base molecule.
In the current version Paratool supports the autogeneration of input
files for Gaussian but later
versions will also provide an interface to
GAMESS.
(For this task Paratool uses
QMtool
which you can also use stand-alone to setup and analyze QM simulations.)
You can obtain the respective interface through the "File->Setup QM optimization". The meaning of the buttons can be found here. In most cases the default values provided by Paratool should be fine, but it is important that you check the total charge and the multiplicity since these are difficult or impossible to guess automatically. a good choice for the simulation method and the basis set are RHF/6-31G* and UB3LYP/6-31+G* for metal containing molecules.
After the Gausian setup file has been written you have to run it yourself. When you load the resulting logfile, the last frame denoting the optimized structure will be appended to the base molecule which makes it possible to easily switch the view between the original and the optimized geometry. Additionally, a new molecule containing all optimization steps, will be loaded in VMD. It will not be shown automatically but you can enable it by hand in the VMD Main window.
...
E(system+water) - E(system) - E(water) ---------------- = E(interaction)Before comparision the QM interaction energy should be scaled with the empirical factor of 1.16. At the same time the force field based optimal distance should be 0.2A shorter that the HF/6-31G* value. The hydration sites are all polar subgroups of the model compound. One typically sets the charges of the direct interaction partner and its mother atom(s). Further all nonpolar hydrogens should obtain a charge of 0.09e. If your model compound is designed properly there will be carbons to use to "neutralize" the entire charge; this could be aliphatic or aromatic carbons that are not involved in significant hydrogen bonding.