



Next: Additional Simulation Parameters
Up: Basic Simulation Parameters
Previous: Full electrostatic integration
Contents
Index
Subsections
NAMD configuration parameters
- cutoff
 local interaction distance common to both electrostatic
and van der Waals calculations (Å)
local interaction distance common to both electrostatic
and van der Waals calculations (Å) 
Acceptable Values: positive decimal
Description: See Section 5.1 for more information.
- switching use switching function?
Acceptable Values: on or off
Default Value: off
Description: If switching is
specified to be off, then a truncated cutoff is performed.
If switching is turned on, then smoothing functions
are applied to both the electrostatics and van der Waals forces.
For a complete description of the non-bonded force parameters see
Section 5.1. If switching is set to
on, then switchdist must also be defined.
- switchdist distance at which to activate switching function
for electrostatic and van der Waals calculations (Å)
Acceptable Values: positive decimal 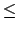 cutoff
cutoff
Description: Distance at which the switching function
should begin to take effect.
This parameter only has meaning if switching is
set to on.
The value of switchdist must be less than
or equal to the value of cutoff, since the switching function
is only applied on the range from switchdist to cutoff.
For a complete description of the non-bonded force parameters see
Section 5.1.
- limitdist maximum distance between pairs for limiting interaction strength(Å)
Acceptable Values: non-negative decimal
Default Value: 0.
Description:
The electrostatic and van der Waals potential functions diverge
as the distance between two atoms approaches zero.
The potential for atoms closer than limitdist is instead
treated as 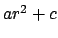 with parameters chosen to match the
force and potential at limitdist.
This option should primarily be useful for alchemical free energy
perturbation calculations, since it makes the process of creating
and destroying atoms far less drastic energetically.
The larger the value of limitdist the more the maximum force
between atoms will be reduced.
In order to not alter the other interactions in the simulation,
limitdist should be less than the closest approach
of any non-bonded pair of atoms; 1.3Å appears to satisfy this
for typical simulations but the user is encouraged to experiment.
There should be no performance impact from enabling this feature.
with parameters chosen to match the
force and potential at limitdist.
This option should primarily be useful for alchemical free energy
perturbation calculations, since it makes the process of creating
and destroying atoms far less drastic energetically.
The larger the value of limitdist the more the maximum force
between atoms will be reduced.
In order to not alter the other interactions in the simulation,
limitdist should be less than the closest approach
of any non-bonded pair of atoms; 1.3Å appears to satisfy this
for typical simulations but the user is encouraged to experiment.
There should be no performance impact from enabling this feature.
- pairlistdist distance between pairs for inclusion in pair lists (Å)
Acceptable Values: positive decimal 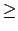 cutoff
cutoff
Default Value: cutoff
Description:
A pair list is generated pairlistsPerCycle times each cycle,
containing pairs of atoms for which
electrostatics and van der Waals interactions will be calculated.
This parameter is used when switching is set to on to
specify the allowable distance between atoms for inclusion in the
pair list.
This parameter is equivalent to the X-PLOR parameter CUTNb.
If no atom moves more than pairlistdist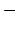 cutoff during
one cycle, then there will be no jump in electrostatic or van der
Waals energies when the next pair list is built. Since such a jump
is unavoidable when truncation is used, this parameter may only
be specified when switching is set to on. If this
parameter is not specified and switching is set to on,
the value of cutoff is used.
A value of at least one greater than cutoff is recommended.
cutoff during
one cycle, then there will be no jump in electrostatic or van der
Waals energies when the next pair list is built. Since such a jump
is unavoidable when truncation is used, this parameter may only
be specified when switching is set to on. If this
parameter is not specified and switching is set to on,
the value of cutoff is used.
A value of at least one greater than cutoff is recommended.
- splitPatch how to assign atoms to patches
Acceptable Values: position or hydrogen
Default Value: hydrogen
Description:
When set to hydrogen, hydrogen atoms are kept on the same patch as their parents, allowing faster distance checking and rigid bonds.
- hgroupCutoff (Å) used for group-based distance testing
Acceptable Values: positive decimal
Default Value: 2.5
Description:
This should be set to twice the largest distance which will ever occur between a hydrogen atom and its mother. Warnings will be printed if this is not the case. This value is also added to the margin.
- margin extra length in patch dimension (Å)
Acceptable Values: positive decimal
Default Value: 0.0
Description: An internal tuning parameter used in determining the size of the cubes
of space with which NAMD uses to partition the system. The value of
this parameter will not change the physical results of the simulation.
Unless you are very motivated to get the very best
possible performance, just leave this value at the default.
- pairlistMinProcs min procs for pairlists
Acceptable Values: positive integer
Default Value: 1
Description:
Pairlists may consume a large amount of memory as atom counts, densities,
and cutoff distances increase. Since this data is distributed across
processors it is normally only problematic for small processor counts.
Set pairlistMinProcs to the smallest number of processors on which
the simulation can fit into memory when pairlists are used.
- pairlistsPerCycle regenerate x times per cycle
Acceptable Values: positive integer
Default Value: 2
Description:
Rather than only regenerating the pairlist at the beginning of a cycle,
regenerate multiple times in order to better balance the costs of
atom migration, pairlist generation, and larger pairlists.
- outputPairlists how often to print warnings
Acceptable Values: non-negative integer
Default Value: 0
Description:
If an atom moves further than the pairlist tolerance during a simulation
(initially (pairlistdist - cutoff)/2 but refined during the run) any
pairlists covering that atom are invalidated and temporary pairlists
are used until the next full pairlist regeneration. All interactions
are calculated correctly, but efficiency may be degraded. Enabling
outputPairlists will summarize these pairlist violation warnings
periodically during the run.
- pairlistShrink tol *= (1 - x) on regeneration
Acceptable Values: non-negative decimal
Default Value: 0.01
Description:
In order to maintain validity for the pairlist for an entire cycle,
the pairlist tolerance (the distance an atom can move without causing
the pairlist to be invalidated) is adjusted during the simulation.
Every time pairlists are regenerated the tolerance is reduced by
this fraction.
- pairlistGrow tol *= (1 + x) on trigger
Acceptable Values: non-negative decimal
Default Value: 0.01
Description:
In order to maintain validity for the pairlist for an entire cycle,
the pairlist tolerance (the distance an atom can move without causing
the pairlist to be invalidated) is adjusted during the simulation.
Every time an atom exceeds a trigger criterion that is some fraction
of the tolerance distance, the tolerance is increased by this fraction.
- pairlistTrigger trigger is atom beyond (1 - x) * tol
Acceptable Values: non-negative decimal
Default Value: 0.3
Description:
The goal of pairlist tolerance adjustment is to make pairlist invalidations
rare while keeping the tolerance as small as possible for best performance.
Rather than monitoring the (very rare) case where atoms actually move more
than the tolerance distance, we reduce the trigger tolerance by this
fraction. The tolerance is increased whenever the trigger tolerance is
exceeded, as specified by pairlistGrow.
- exclude exclusion policy to use
Acceptable Values: none, 1-2, 1-3, 1-4, or scaled1-4
Description: This parameter specifies which pairs of bonded atoms should
be excluded from non-bonded
interactions. With the value of none, no bonded pairs of atoms
will be excluded. With the value of 1-2, all atom pairs that
are directly connected via a linear bond will be excluded. With the
value of 1-3, all 1-2 pairs will be excluded along with
all pairs of atoms that are bonded to a common
third atom (i.e., if atom A is bonded to atom B and atom B is bonded
to atom C, then the atom pair A-C would be excluded).
With the value of 1-4, all 1-3 pairs will be excluded along
with all pairs connected by a set of two bonds (i.e., if atom A is bonded
to atom B, and atom B is bonded to atom C, and atom C is bonded to
atom D, then the atom pair A-D would be excluded). With the value
of scaled1-4, all 1-3 pairs are excluded and all pairs
that match the 1-4 criteria are modified. The electrostatic
interactions for such pairs are modified by the constant factor
defined by 1-4scaling.
The van der Waals interactions are modified
by using the special 1-4 parameters defined in the parameter files.
- temperature initial temperature (K)
Acceptable Values: positive decimal
Description: Initial temperature value for the system.
Using this option will generate a random
velocity distribution for the initial velocities
for all the atoms such that the system
is at the desired temperature.
Either the temperature
or the velocities/binvelocities
option must be defined to determine an initial set of velocities.
Both options cannot be used together.
- COMmotion allow initial center of mass motion?
Acceptable Values: yes or no
Default Value: no
Description:
Specifies whether or not motion of
the center of mass of the entire system is allowed.
If this option is set to no, the initial velocities of the system
will be adjusted to remove center of mass motion of the system.
Note that this does not preclude later center-of-mass motion due to
external forces such as random noise in Langevin dynamics, boundary
potentials, and harmonic restraints.
- zeroMomentum remove center of mass drift due to PME
Acceptable Values: yes or no
Default Value: no
Description:
If enabled, the net momentum of the simulation and any resultant drift
is removed before every full electrostatics step.
This correction should conserve energy and have minimal impact on
parallel scaling.
This feature should only be used for simulations that would
conserve momentum except for the slight errors in PME.
(Features such as fixed atoms, harmonic restraints, steering forces,
and Langevin dynamics do not conserve momentum; use in combination
with these features should be considered experimental.)
Since the momentum correction is delayed, enabling outputMomenta
will show a slight nonzero linear momentum but there should be no
center of mass drift.
- dielectric dielectric constant for system
Acceptable Values: decimal 1.0
Default Value: 1.0
Description: Dielectric constant for the system. A value of 1.0 implies no modification
of the electrostatic interactions. Any larger value will lessen the
electrostatic forces acting in the system.
- 1-4scaling scaling factor for 1-4 interactions
Acceptable Values: 0 decimal 1
Default Value: 1.0
Description: Scaling factor for 1-4 interactions. This factor is only used when the
exclude parameter is set to scaled1-4. In this case, this
factor is used to modify the electrostatic interactions between 1-4 atom
pairs. If the exclude parameter is set to anything but
scaled1-4, this parameter has no effect regardless of its value.
- vdwGeometricSigma use geometric mean to combine L-J sigmas
Acceptable Values: yes or no
Default Value: no
Description: Use geometric mean, as required by OPLS, rather than
traditional arithmetic mean when combining Lennard-Jones sigma parameters
for different atom types.
- seed random number seed
Acceptable Values: positive integer
Default Value: pseudo-random value based on current UNIX clock time
Description: Number used to seed the random number generator
if temperature or langevin is selected. This can be
used so that consecutive simulations produce the same results.
If no value is specified, NAMD will choose a pseudo-random
value based on the current UNIX clock time. The random number
seed will be output during the simulation startup so that
its value is known and can be reused for subsequent simulations.
Note that if Langevin dynamics are used in a parallel simulation
(i.e., a simulation using more than one processor)
even using the same seed will not guarantee reproducible results.
- rigidBonds controls if and how ShakeH is used
Acceptable Values: none,
water, all
Default Value: none
Description: When water is selected, the hydrogen-oxygen and hydrogen-hydrogen
distances in waters are constrained to the nominal length or angle given
in the parameter file, making the molecules completely rigid.
When rigidBonds is all, waters are made rigid as described above
and the bond between each hydrogen and the (one) atom to which
it is bonded is similarly constrained.
For the default case none, no lengths are constrained.
- rigidTolerance allowable bond-length error for ShakeH (Å)
Acceptable Values: positive decimal
Default Value: 1.0e-8
Description:
The ShakeH algorithm is assumed to have converged when all constrained
bonds differ from the nominal bond length by less than this amount.
- rigidIterations maximum ShakeH iterations
Acceptable Values: positive integer
Default Value: 100
Description:
The maximum number of iterations ShakeH will perform before giving up
on constraining the bond lengths. If the bond lengths do not
converge, a warning message is printed, and the atoms are left at the
final value achieved by ShakeH.
Although the default value is 100,
convergence is usually reached after fewer than 10 iterations.
- rigidDieOnError maximum ShakeH iterations
Acceptable Values: on or off
Default Value: on
Description:
Exit and report an error if rigidTolerance is not achieved after rigidIterations.
- useSettle Use SETTLE for waters.
Acceptable Values: on or off
Default Value: on
Description:
If rigidBonds are enabled then use the non-iterative SETTLE algorithm to
keep waters rigid rather than the slower SHAKE algorithm.
DPMTA is no longer included in the released NAMD binaries.
We recommend that you instead use PME with a periodic system because
it conserves energy better, is more efficient, and is better parallelized.
If you must have the fast multipole algorithm you may compile NAMD yourself.
These parameters control the options to DPMTA, an algorithm
used to provide full electrostatic interactions. DPMTA is a
modified version of the FMA (Fast Multipole Algorithm) and,
unfortunately, most of the parameters still refer to FMA
rather than DPMTA for historical reasons. Don't be confused!
For a further description of how exactly full electrostatics
are incorporated into NAMD, see Section 5.2.
For a greater level of detail about DPMTA and the specific
meaning of its options, see the DPMTA distribution which is
available via anonymous FTP from the site ftp.ee.duke.edu
in the directory /pub/SciComp/src.
PME stands for Particle Mesh Ewald and is an efficient
full electrostatics method for use with periodic boundary conditions.
None of the parameters should affect energy conservation, although they may affect the accuracy of the results and momentum conservation.
- PME Use particle mesh Ewald for electrostatics?
Acceptable Values: yes or no
Default Value: no
Description: Turns on particle mesh Ewald.
- PMETolerance PME direct space tolerance
Acceptable Values: positive decimal
Default Value: 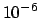
Description: Affects the value of the Ewald coefficient and the overall accuracy of the results.
- PMEInterpOrder PME interpolation order
Acceptable Values: positive integer
Default Value: 4 (cubic)
Description: Charges are interpolated onto the grid and forces are interpolated off using this many points, equal to the order of the interpolation function plus one.
- PMEGridSpacing maximum space between grid points
Acceptable Values: positive real
Description: The grid spacing partially determines the accuracy and efficiency of PME.
If any of the grid sizes below are not set, then PMEGridSpacing must be set
(recommended value is 1.0 Å) and will be used to calculate them.
If a grid size is set, then the grid spacing must be
at least PMEGridSpacing (if set, or a very large default of 1.5).
- PMEGridSizeX number of grid points in x dimension
Acceptable Values: positive integer
Description: The grid size partially determines the accuracy and efficiency of PME.
For speed, PMEGridSizeX should have only small integer factors (2, 3 and 5).
- PMEGridSizeY number of grid points in y dimension
Acceptable Values: positive integer
Description: The grid size partially determines the accuracy and efficiency of PME.
For speed, PMEGridSizeY should have only small integer factors (2, 3 and 5).
- PMEGridSizeZ number of grid points in z dimension
Acceptable Values: positive integer
Description: The grid size partially determines the accuracy and efficiency of PME.
For speed, PMEGridSizeZ should have only small integer factors (2, 3 and 5).
- PMEProcessors processors for FFT and reciprocal sum
Acceptable Values: positive integer
Default Value: larger of x and y grid sizes up to all available processors
Description: For best performance on some systems and machines, it may be necessary to
restrict the amount of parallelism used. Experiment with this parameter if
your parallel performance is poor when PME is used.
- FFTWEstimate Use estimates to optimize FFT?
Acceptable Values: yes or no
Default Value: no
Description: Do not optimize FFT based on measurements, but on FFTW rules of thumb.
This reduces startup time, but may affect performance.
- FFTWUseWisdom Use FFTW wisdom archive file?
Acceptable Values: yes or no
Default Value: yes
Description: Try to reduce startup time when possible by reading FFTW ``wisdom'' from a file, and saving wisdom generated by performance measurements to the same file for future use.
This will reduce startup time when running the same size PME grid on the same number of processors as a previous run using the same file.
- FFTWWisdomFile name of file for FFTW wisdom archive
Acceptable Values: file name
Default Value: FFTW_NAMD_version_platform.txt
Description: File where FFTW wisdom is read and saved.
If you only run on one platform this may be useful to reduce startup times for all runs.
The default is likely sufficient, as it is version and platform specific.
- useDPME Use old DPME code?
Acceptable Values: yes or no
Default Value: no
Description: Switches to old DPME implementation of particle mesh Ewald.
The new code is faster and allows non-orthogonal cells so you
probably just want to leave this option turned off. If you set
cellOrigin to something other than 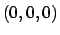 the energy may differ
slightly between the old and new implementations.
DPME is no longer included in released binaries.
the energy may differ
slightly between the old and new implementations.
DPME is no longer included in released binaries.
The direct computation of electrostatics
is not intended to be used during
real calculations, but rather as a testing or
comparison measure. Because of the
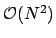 computational complexity for performing
direct calculations, this is much
slower than using DPMTA or PME to compute full
electrostatics for large systems.
In the case of periodic boundary conditions,
the nearest image convention is used rather than a
full Ewald sum.
computational complexity for performing
direct calculations, this is much
slower than using DPMTA or PME to compute full
electrostatics for large systems.
In the case of periodic boundary conditions,
the nearest image convention is used rather than a
full Ewald sum.
- FullDirect calculate full electrostatics directly?
Acceptable Values: yes or no
Default Value: no
Description: Specifies whether or not direct computation of
full electrostatics should be performed.
One of the areas of current research being studied using NAMD is the
exploration of better methods for performing multiple timestep integration.
Currently the only available method is the impulse-based Verlet-I or r-RESPA
method which is stable for timesteps up to 4 fs for long-range electrostatic
forces, 2 fs for short-range nonbonded forces, and 1 fs for bonded forces
Setting rigid all (i.e., using SHAKE) increases these timesteps to
6 fs, 2 fs, and 2 fs respectively but eliminates bond motion for hydrogen.
The mollified impulse method (MOLLY) reduces the resonance which limits
the timesteps and thus increases these timesteps to 6 fs, 2 fs, and 1 fs
while retaining all bond motion.
Next: Additional Simulation Parameters
Up: Basic Simulation Parameters
Previous: Full electrostatic integration
Contents
Index
namd@ks.uiuc.edu