



Next: Index
Up: NAMD 2.6 User's Guide
Previous: Documentation
Contents
Index
- 1
-
M. P. Allen and D. J. Tildesley.
Computer Simulation of Liquids.
Oxford University Press, New York, 1987.
- 2
-
P. H. Axelsen and D. Li.
Improved convergence in dual-topology free energy calculations
through use of harmonic restraints.
J. Comput. Chem., 19:1278-1283, 1998.
- 3
-
F. C. Bernstein, T. F. Koetzle, G. J. B. Williams, J. E. F. Meyer, M. D. Brice,
J. R. Rodgers, O. Kennard, T. Shimanouchi, and M. Tasumi.
The protein data bank: A computer-based archival file for
macromolecular structures.
J. Mol. Biol., 112:535-542, 1977.
- 4
-
D. L. Beveridge and F. M. DiCapua.
Free energy via molecular simulation: applications to chemical and
biomolecualr systems.
Ann. Rev. Biophys. Biophys. Chem., 18:431-492, 1989.
- 5
-
S. Boresch and M. Karplus.
The role of bonded terms in free energy simulations. 1. theoretical
analysis.
J. Phys. Chem. A, 103:103-118, 1999.
- 6
-
B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan,
and M. Karplus.
CHARMM: a program for macromolecular energy, minimization, and
dynamics calculations.
J. Comp. Chem., 4(2):187-217, 1983.
- 7
-
A. T. Brünger.
X-PLOR, Version 3.1, A System for X-ray Crystallography and
NMR.
The Howard Hughes Medical Institute and Department of Molecular
Biophysics and Biochemistry, Yale University, 1992.
- 8
-
D. Chandler.
Introduction to modern statistical mechanics.
Oxford University Press, 1987.
- 9
-
C. Chipot and D. A. Pearlman.
Free energy calculations. the long and winding gilded road.
Mol. Sim., 2001.
- 10
-
E. Darve and A. Pohorille.
Calculating free energies using average force.
J. Chem. Phys., 115:9169-9183, 2001.
- 11
-
W. K. den Otter and W. J. Briels.
Free energy from molecular dynamics with multiple constraints.
Mol. Phys., 98:773-781, 2000.
- 12
-
M. K. Gilson, J. A. Given, B. L. Bush, and J. A. McCammon.
The statistical-thermodynamic basis for computation of binding
affinities: a critical review.
Biophys. J., 72:1047-1069, 1997.
- 13
-
J. Hénin and C. Chipot.
Overcoming free energy barriers using unconstrained molecular
dynamics simulations.
J. Chem. Phys., 121:2904-2914, 2004.
- 14
-
W. Humphrey and A. Dalke.
VMD user guide (Version 0.94).
Beckman Institute Technical Report TB-94-07, University of Illinois,
1994.
- 15
-
P. M. King.
Free energy via molecular simulation: A primer.
In W. F. van Gunsteren, P. K. Weiner, and A. J. Wilkinson, editors,
Computer simulation of biomolecular systems: Theoretical and
experimental applications, volume 2, pages 267-314. ESCOM, Leiden, 1993.
- 16
-
P. A. Kollman.
Free energy calculations: applications to chemical and biochemical
phenomena.
Chem. Rev., 93:2395-2417, 1993.
- 17
-
A. E. Mark.
Free energy perturbation calculations.
In P. Schleyer, editor, Encyclopaedia of computational
chemistry, volume 2, pages 1070-1083. John Wiley and Sons, New York, 1998.
- 18
-
J. A. McCammon and S. C. Harvey.
Dynamics of Proteins and Nucleic Acids.
Cambridge University Press, Cambridge, 1987.
- 19
-
D. A. Pearlman.
A comparison of alternative approaches to free energy calculations.
J. Phys. Chem., 98:1487-1493, 1994.
- 20
-
D. Rodriguez-Gomez, E. Darve, and A. Pohorille.
Assessing the efficiency of free energy calculation methods.
J. Chem. Phys., 120:3563-3578, 2004.
- 21
-
A. Roitberg and R. Elber.
Modeling side chains in peptides and proteins: Application of the
locally enhanced sampling technique and the simulated annealing methods to
find minimum energy conformations.
95:9277-9287, 1991.
- 22
-
C. Simmerling, T. Fox, and P. A. Kollman.
Use of locally enhanced sampling in free energy calculations:
Testing and application to the
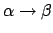 anomerization of
glucose.
anomerization of
glucose.
120(23):5771-5782, 1998.
- 23
-
C. Simmerling, M. R. Lee, A. R. Ortiz, A. Kolinski, J. Skolnick, and P. A.
Kollman.
Combining MONSSTER and LES/PME to predict protein structure from
amino acid sequence: Application to the small protein CMTI-1.
122(35):8392-8402, 2000.
- 24
-
M. Sprik and G. Cicotti.
Free energy from constrained molecular dynamics.
J. Chem. Phys., 109:7737-7744, 1998.
- 25
-
T. Straatsma and A. McCammon.
Computational alchemy.
Ann. Rev. Phys. Chem., 43:407-435, 1992.
- 26
-
G. M. Torrie and J. P. Valleau.
Nonphysical sampling distributions in monte carlo free energy
estimation: Umbrella sampling.
J. Comput. Phys., 23:187-199, 1977.
- 27
-
W. F. van Gunsteren.
Methods for calculation of free energies and binding constants:
successes and problems.
In W. F. Van Gunsteren and P. K. Weiner, editors, Computer
simulation of biomolecular systems: theoretical and experimental
applications, pages 27-59. ESCOM Science Publishers B. V., The Netherlands,
1989.
- 28
-
R. W. Zwanzig.
High temperature equation of state by a perturbation method. i.
nonpolar gases.
J. Chem. Phys., 22:1420-1426, 1954.
namd@ks.uiuc.edu