



Next: Generalized Born Implicit Solvent
Up: Force Field Parameters
Previous: MARTINI Residue-Based Coarse-Grain Forcefield
Contents
Index
Subsections
Bond constraint parameters
The following describes the parameters for the
harmonic restraints feature of NAMD.
For historical reasons the terminology of
``harmonic constraints'' has been carried over from X-PLOR.
This feature allows a harmonic restraining force to be applied
to any set of atoms in the simulation.
- constraints
 are constraints active?
are constraints active? 
Acceptable Values: on or off
Default Value: off
Description: Specifies whether or not harmonic constraints are active. If it
is set to off, then no harmonic constraints are computed.
If it is set to on, then
harmonic constraints are calculated using the values specified
by the parameters consref, conskfile, conskcol,
and consexp.
- consexp
exponent for harmonic constraint energy function
Acceptable Values: positive, even integer
Default Value: 2
Description: Exponent to be use in the harmonic constraint energy function.
This value must be a positive integer, and only even values really make
sense. This parameter is used only if constraints is set to
on.
- consref
PDB file containing constraint reference positions
Acceptable Values: UNIX file name
Description: PDB file to use for reference positions for harmonic constraints.
Each atom that has an active constraint will be constrained about
the position specified in this file.
- conskfile
PDB file containing force constant values
Acceptable Values: UNIX filename
Description: PDB file to use for force constants for
harmonic constraints.
- conskcol
column of PDB file containing force constant
Acceptable Values: X, Y, Z, O, or B
Description: Column of the PDB file to use for the harmonic constraint force constant.
This parameter may specify any of the floating point fields of the PDB file,
either X, Y, Z, occupancy, or beta-coupling (temperature-coupling).
Regardless of which column is used, a value of 0 indicates that the atom
should not be constrained.
Otherwise, the value specified is used as the force constant for
that atom's restraining potential.
- constraintScaling
scaling factor for harmonic constraint energy function
Acceptable Values: positive
Default Value: 1.0
Description: The harmonic constraint energy function is multiplied by this parameter,
making it possible to gradually turn off constraints during equilibration.
This parameter is used only if constraints is set to
on.
- selectConstraints
Restrain only selected Cartesian components of the coordinates?
Acceptable Values: on or off
Default Value: off
Description: This option is useful to restrain the positions of atoms to a plane or a line in space. If active,
this option will ensure that only selected Cartesian components of the coordinates are restrained.
E.g.: Restraining the positions of atoms to their current z values with no restraints
in x and y will allow the atoms to move in the x-y plane while retaining their original z-coordinate.
Restraining the x and y values will lead to free motion only along the z coordinate.
- selectConstrX
Restrain X components of coordinates
Acceptable Values: on or off
Default Value: off
Description: Restrain the Cartesian x components of the positions.
- selectConstrY
Restrain Y components of coordinates
Acceptable Values: on or off
Default Value: off
Description: Restrain the Cartesian y components of the positions.
- selectConstrZ
Restrain Z components of coordinates
Acceptable Values: on or off
Default Value: off
Description: Restrain the Cartesian z components of the positions.
Atoms may be held fixed during a simulation. NAMD avoids calculating most interactions in which all affected atoms are fixed unless fixedAtomsForces is specified.
Additional bond, angle, and dihedral energy terms may be applied to system,
allowing secondary or tertiary structure to be restrained, for example.
Extra bonded terms are not considered part of the molecular structure
and hence do not alter nonbonded exclusions.
The energies from extra bonded terms are included with the normal
bond, angle, and dihedral energies in NAMD output.
All extra bonded terms are harmonic potentials of the form
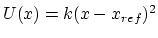 except dihedrals and impropers with
a non-zero periodicity specified, which use
except dihedrals and impropers with
a non-zero periodicity specified, which use
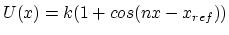 .
The only difference between dihedrals and
impropers is the output field that their potential energy is added to.
.
The only difference between dihedrals and
impropers is the output field that their potential energy is added to.
The extra bonded term implementation shares the parallel implementation
of regular bonded terms in NAMD, allowing large numbers of extra terms
to be specified with minimal impact on parallel scalability.
Extra bonded terms do not have to duplicate normal bonds/angles/dihedrals,
but each extra bond/angle/dihedral should only involve nearby atoms.
If the atoms involved are too far apart a bad global bond count will be
reported in parallel runs.
Extra bonded terms are enabled via the following options:
The extra bonds file(s) should contain lines of the following formats:
- bond <atom> <atom> <k> <ref>
- angle <atom> <atom> <atom> <k> <ref>
- dihedral <atom> <atom> <atom> <atom> <k> <ref>
- dihedral <atom> <atom> <atom> <atom> <k> <n> <ref>
- improper <atom> <atom> <atom> <atom> <k> <ref>
- improper <atom> <atom> <atom> <atom> <k> <n> <ref>
- # <comment ...>
In all cases <atom> is a zero-based atom index
(the first atom has index 0),
<ref> is a reference distance in Å (bond) or angle in degrees (others),
and <k> is a spring constant in the potential energy function
or, for dihedrals and impropers with
periodicity <n> specified and not 0,
.
Note that 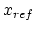 is only a minimum for the harmonic potential;
the sinusoidal potential has minima at
is only a minimum for the harmonic potential;
the sinusoidal potential has minima at
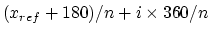 .
.
Next: Generalized Born Implicit Solvent
Up: Force Field Parameters
Previous: MARTINI Residue-Based Coarse-Grain Forcefield
Contents
Index
http://www.ks.uiuc.edu/Research/namd/