


Next: Acknowledgements
Up: Aquaporin Tutorial
Previous: Phylogenetic Tree
Contents
Subsections
Evolutionary Profile of AQPs
So far you have learned how to construct a phylogenetic tree using the structural alignment of AQPs.
The sequence alignment can also be used to build a phylogenetic tree, especially when protein structures are not
available.
To make use of both the structural and sequence information,
Multiseq now allows you to merge the two types of alignments and construct a complete
evolutionary profile (EP) for the proteins being studied. In this section,
you will learn how to obtain the EP for AQPs. For more information on using Multiseq
program to perform evolutionary analysis, please refer to the Evolution of Biomolecular Structure tutorial.
For the following section you will
need to install BLAST on your computer. BLAST is a software that searches
through sequence databases and locate those sequences that are similar to a
query sequence. It is available online at http://www.ncbi.nlm.nih.gov/BLAST/ (click on Help tab, find and click the
Download link. At the bottom of the section titled "Legacy executables," click
on the link ftp://ftp.ncbi.nlm.nih.gov/blast/executables/release/LATEST/,
and download BLAST as a guest). Here we will install a local copy of BLAST for
Multiseq.
- 1
- Create a directory into which BLAST will be installed.
Examples:
Unix/Linux:/usr/local/blast;
Mac OS X:/Applications/Blast;
Windows: C: Blast
Blast
- 2
- Extract the archives of BLAST.
Copy the blast installation file for your platform from the aqp-tutorial-files  blast-install directory
to the directory you've made. In Unix or Linux, extract the files by using
the command tar zxvf filename. On Mac OS X or Windows, double-click the file.
blast-install directory
to the directory you've made. In Unix or Linux, extract the files by using
the command tar zxvf filename. On Mac OS X or Windows, double-click the file.
Figure:
Choose the directory for BLAST. The final directory in the BLAST Installation Directory should now be titled blast-2.2.26 as the BLAST version has changed.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=3 in]{pictures/blastinstall}
}
\end{center}\vspace{-0.7cm}\end{figure}](img61.gif) |
- 3
- Do the same for swiss-prot.tar.gz.
Repeat the above two steps: create a directory for swiss-prot,
copy the file swiss-prot.tar.gz from aqp-tutorial-files to the directory
you've created, and extract it.
- 4
- Set the BLAST installation location in Multiseq.
In the Multiseq program window, choose File Preferences.
Click on the Software button in the new dialog to bring up the software preferences.
Click on the Browse button in the BLAST Installation Directory section and select
the directory into which you installed BLAST (Fig. 33).
Note: You may be asked by Multiseq to update certain databases before you could continue,
if so, click Yes and wait for Multiseq to finish the update.
In Linux or Mac OS X, you may have a directory named blast-2.2.26
in your installation directory. Pick this directory if you have it.
AQPs are present in all three domains of life. To build a complete EP for AQPs,
we will first perform a structural alignment for AQPs in all three domains of life: Eukaryota,
Bacteria, and Archaea. In the previous sections, you have seen the structures of human AQP1 (1fqy) and
E .coli AqpZ (1rc2). Here you will also need AqpM (2f2b) from Archaea to construct the EP.
We have provided these pdb files in the tutorial files for you.
Figure:
Import structures for AQPs in Multiseq.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=3 in]{pictures/importfromblast}
}
\end{center}\vspace{-0.7cm}\end{figure}](img62.gif) |
- 1
- Open a new VMD and load the pdb files 1fqy, 1rc2, and 2f2b one by one.
- 2
- Open the Multiseq program by clicking Extensions Analysis Multiseq.
- 3
- In the Multiseq program window, keep the protein structures under VMD Protein Structures and delete
all structures under VMD Nucleic Structures.
If you loaded your structures by giving the pdb code to VMD (not with the files we prepared for you), you may have more than one structure for each of
the pdb code you entered, i.e., besides 1rc2A, you may also have 1rc2B. This indicates that in the original pdb file,
there are two different structures for the protein. The difference between them is usually very small, and does not affect
the alignment we are going to perform. Therefore, simply delete 1rc2B and keep 1rc2A.
Now that you have the structures of AQPs loaded, we will use BLAST to find sequences of AQPs in all three domains of life.
Each of the three structures will be used as a query sequence by BLAST, and sequences in the swiss-prot database will be compared
with them, one at a time. Those sequences similar to our query sequence will be picked by BLAST and loaded in Multiseq.
- 1
- In the Multiseq window, check the box in front of 1fqy. Then click File Import Data.
You will find the same window you've seen when loading the pdb structures. This time, choose From BLAST Search under Data Source and select Marked Sequences (Fig. 34).
- 2
- Click the Browse button after Databases, and go to the directory where you extracted the file swiss-prot.tar.gz. You should find a direcotry named swiss-prot. Go into that directory and select the file uniprotsprot.
- 3
- Choose
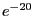 for E Score and 1 for Iterations and then click OK.
for E Score and 1 for Iterations and then click OK.
BLAST is now searching the database with 1fqy as a query sequence. This should take a minute or two.
A new window named BLAST Search Results will open once the search has finished. Note that the swiss-prot database provided here only contains sequence data for proteins in this session. You cannot rely on it for other proteins that you want to investigate. Moreover, the database is not an updated one, so visit the BLAST online databases if you want the latest results.
As you may have noticed, 100 sequences have been found using the query sequence 1fqy. We will only keep those sequences from
the Eukaryota domain, since our query sequence is from Eukaryota. Later we will find sequences in Bacteria and Archaea using the query sequences
1rc2 and 2f2b, respectively. This should make our search more accurate.
- 4
- In the BLAST Search Results window, under Domains, unselect the All list and select Eukaryota. Click Apply Filter.
You will find that only 87 sequences are left (Fig. 35).
- 5
- Click Accept. The sequences will be loaded in Multiseq.
Figure:
Search result of BLAST.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=3 in]{pictures/blastresult}
}
\end{center}\vspace{-0.7cm}\end{figure}](img64.gif) |
- 6
- Check the box in front of 1rc2 and uncheck 1fqy in the Multiseq window.
Now you could repeat the above process and find Bacteria sequences using 1rc2 as a query sequence. You should find 28 sequences from Bacteria.
Repeat this process using 2f2b as a query sequence and get 3 sequences for Archaea.
Before we continue, save your Multiseq session by clicking File Save Session and save it as aqp.multiseq.
You can load the session later by clicking clicking File Load Session.
There is a saved aqp.multiseq session in the tutorial files, in case you'd like to check with it.
In order to analyze the three structures and the 118 sequences of AQPs together, we need to first align them.
What we will do is to first align the structures using the STAMP structural alignment tool mentioned in section 2, and
then we will use the structural alignment to guide the sequence alignments.
- 1
- Mark the three pdb structures by checking the boxes in front of them. Make sure that no other sequences are marked.
- 2
- Click Tools Stamp Structural Alignment and choose to align Marked Structures and then click OK.
- 3
- Unmark structures and mark all the sequences. Remove gaps in the sequences by clicking Edit Remove Gaps and then select Remove gaps from: Marked sequences, and Remove these types of gaps: All gaps.
You could select all the sequences at once by clicking on the first sequence, pressing the shift button and then clicking on the last sequence.
All the sequences should appear in yellow now, which means they are highlighted. Press the shift button and check one box in front of any highlighted sequence. All other boxes for the highlighted sequences should be automatically checked.
- 4
- Highlight all sequences and all structures as described above, so that
all sequences appear yellow and all boxes in front of sequence identifiers are
checked, then click Tools Sequence Alignment.
A new
window named Sequence Alignment Options should appear
(Fig 36). Check ClustalW under Alignment
Program. As we are going to align the sequences using the structural
alignment, choose Profile/Sequence Alignment instead of Multiple
Alignment in the window. Under Align marked sequences to group, select
VMD Protein Structures, and then click OK. This should take
two or three minutes.
Figure:
Sequence Alignment Options window.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=2 in]{pictures/clustalw}
}
\end{center}\vspace{-0.7cm}\end{figure}](img65.gif) |
Now you have a complete structural based alignment of the AQPs in all three domains. Try coloring it by sequence identity by
clicking View Coloring Sequence Identity (Fig.37).
Figure:
ClustalW alignment result using the structural profile
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=3.5 in]{pictures/alignwstrucprofile}
}
\end{center}\vspace{-0.7cm}\end{figure}](img66.gif) |
Although we have obtained the structures and sequences for AQPs in all three domains and aligned them together, what we have now is not
an evolutionary profile yet. We still need to get rid of the redundancy in these sequences caused by the biased databases.
Multiseq provides a Sequence QR tool which can be used to select a minimum non-redundant set from the sequences, using
a threshold specified by the user.
- 1
- Mark all the sequences and make sure that the structures are unmarked. Click Search Select Non-Redundant Set from the menu.
A new window named Select Non-Redundant Set should show up (Fig. 38). In this window, choose Select from Marked Sequences,
and choose Using Sequence QR. Set the Maximum PID to 75 and then click OK.
Figure:
Select Non-Redundant Set window.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=2 in]{pictures/QRfactorization}
}
\end{center}\vspace{-0.7cm}\end{figure}](img67.gif) |
You should find that some of your sequences are highlighted after the program stopped calculating. These represent the non-redundant set that Multiseq selected for you. Group them together by clicking Options Grouping From Selection and enter ``NR set'' for the new group. This should put all your highlighted sequences into a group named ``NR set''. This is the evolutionary profile (EP) for AQPs.
You could now create the phylogenetic tree using the EP of AQPs: simply delete all the sequences except the ones in the NR set and create a phlogenetic tree as you did in section 7.
Evolutionary profile provides an ``unbiased'' view for the evolutionary relationship of the proteins in investigation. Using EP, scientists have successfully
identified a new subfamily for the protein cysteinyl-tRNA synthetase. For more details on constructing EP and performing evolutionary analysis, please refer
to the Evolution of Biomolecular Structure tutorial.
Next: Acknowledgements
Up: Aquaporin Tutorial
Previous: Phylogenetic Tree
Contents
school@ks.uiuc.edu
