

Next: Working with Multiple Molecules
Up: VMD Tutorial
Previous: Scripting in VMD
Subsections
Data Analysis in VMD
VMD is a powerful tool for analysis of structures and trajectories. Numerous tools for analysis are available under the VMD Main menu item Extensions
 Analysis. In addition to these built-in tools, VMD users often use custom-written scripts to analyze desired properties of the simulated systems. VMD Tcl scripting capabilities are very extensive, and provide boundless opportunities for analysis. In this following section, we will learn how to use built-in VMD features for standard analysis, as well as consider a simple example of scripting.
Analysis. In addition to these built-in tools, VMD users often use custom-written scripts to analyze desired properties of the simulated systems. VMD Tcl scripting capabilities are very extensive, and provide boundless opportunities for analysis. In this following section, we will learn how to use built-in VMD features for standard analysis, as well as consider a simple example of scripting.
Labels
Labels can be placed in VMD to get information on a particular selection, to be used during visualization and quantitative analysis. Labels are selected with the mouse and can be accessed in Graphics
Labels menu. We will cover labels that can be placed on atoms and bonds, although angle and dihedral labeling are also available. In this context, labels for ``bonds'' or ``angles'' actually mean distances between two atoms or angles between three atoms; the atoms do not have to be physically connected by bonds in the molecule.
- 1
- Start a new VMD session. Load the ubiquitin trajectory into VMD (using the files ubiquitin.psf and pulling.dcd). For graphical representation, display protein only, using New Cartoon for drawing method and Structure for coloring method. If you need help, check Section 2.1, steps 1-5.
- 2
- Choose the Mouse
Labels
Atoms menu item from the VMD Main menu. The mouse is now set to the mode for displaying atom labels. You can click on any atom on your molecule and a label will be placed for this atom. Clicking again on it will erase the label.
- 3
- We will now try the same for bonds. Choose the Mouse
Label
Bonds menu item from the VMD Main menu. This selects the ``Display Label for Bond'' mode.
We will consider the distance between the 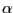 carbon of Lysine 48 and of the C terminus. In the pulling simulation, the former is kept fixed, and the latter is pulled at a constant force of 500 pN. In reality, polyubiquitin chains can be linked by a connection between the C terminus of one ubiquitin molecule and the Lysine 48 of the next. The simulation then mimics the effect of pulling on the C terminus with this kind of linkage.
carbon of Lysine 48 and of the C terminus. In the pulling simulation, the former is kept fixed, and the latter is pulled at a constant force of 500 pN. In reality, polyubiquitin chains can be linked by a connection between the C terminus of one ubiquitin molecule and the Lysine 48 of the next. The simulation then mimics the effect of pulling on the C terminus with this kind of linkage.
- 4
- We will make a VDW representation for the
carbons of Lysine 48 and of the C terminus. To find out the index of these atoms, make a selection including these two atoms, by typing in the Tk Console window (to open the Tk Console window, select Extensions
Tk Console in the VMD Main menu.):
set sel [atomselect top "resid 48 76 and name CA"].
- 5
- Get the indices by typing the following line in the Tk Console window:
$sel get index
This command should give the indices 770 1242.
- 6
- In the Graphical Representations window, create a representation for the selection index 770 1242, with VDW as drawing method.
- 7
- Now that you can see the two
carbons, choose the Mouse
Label
Bonds menu item from the VMD Main menu. Click on each atom one after the other. You should get a line connecting the two atoms (Fig. 24). The number appearing next to the line is the distance between the two atoms in Ångstroms. Note that the appearance of the line (its color), as well as appearance of essentially all other objects in VMD, can be changed in Graphics
Colors in the VMD Main menu.
- 8
- The value of the distance displayed corresponds to the current frame. Try playing the trajectory - you will see that the label is modified automatically as the distance between the atoms changes.
Figure 24:
Labels in VMD. Label control is available in the Graphics
Labels menu item, using which, one can, e.g., plot the labeled distance as a function of
time.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=0.8\textwidth]{FIGS/labels}
}
\end{center}
\end{figure}](img69.gif) |
- 9
- The labels can be used not only for display, but also for obtaining quantitative information. In VMD Main menu, select Graphics
Labels. On the top left-hand side of the window, there is a pull-down menu where you can choose the type of label (Atoms, Bonds, Angles, Dihedrals). For now, keep it in Atoms. You can see the list of atoms for which you have made a label.
- 10
- Click on one of the atoms. You can see all the information of the atom displayed on the bottom half of the Labels window. This information is useful to make selections; it corresponds to the current frame, and is updated as the frame is changed.
- 11
- You can also delete, hide, or show the atom label by clicking on the corresponding button on the top of the Labels window.
- 12
- Now, in the Labels window, choose the label type Bonds, and select the ``bond'' (distance) you labeled (Fig. 24). The information given corresponds to only the first atom in the bond, but the number in the Value field corresponds to the length of the bond in Ångstroms.
- 13
- Click on the Graph tab. Select the bond you labeled between atoms 770 and 1242. Click on the Graph button. This will create a plot of the distance between these two atoms over time (Fig. 24). You can also save this data to a file by clicking on the Save button, and then use an external plotting program to visualize the data.
- 14
- Quit VMD.
Example of a built-in analysis tool: the RMSD Trajectory Tool
The built-in analysis tools in VMD are available under the menu item Extensions
Analysis. These tools each features a GUI window that allow one to enter parameters and
customize the quantities analyzed. In addition, all tools can be invoked in a scripting mode, using the TkConsole window. We will learn how to work with one of the most frequently
used tools, the RMSD Trajectory Tool.
In this example, we will analyze RMSD for two trajectories for the same system, ubiquitin.psf. One of them is the already familiar pulling trajectory, pulling.dcd, and the other is the trajectory of a simulation in which no force was applied to the protein, equilibration.dcd.
- 1
- Start a new VMD session. Load the ubiquitin equilibration trajectory into VMD (using the files ubiquitin.psf and equilibration.dcd).
- 2
- Choose Extensions
Analysis
RMSD Trajectory Tool in the VMD Main window (Fig. 25). The RMSD Trajectory Tool window will show up.
- 3
- In the RMSD Trajectory Tool window, you can see many customizations can be made. For the default values, the molecule to be analyzed is ubiquitin.psf (the only one loaded). The selection for which RMSD will be computed is all of the protein atoms, excluding hydrogens (since thenoh checkbox is on). The RMSD will be calculated for each frame (time
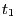 in Eq. 1) with the reference to frame 0 (time
in Eq. 1) with the reference to frame 0 (time 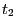 in Eq. 1). Make sure the Plot checkbox is selected.
in Eq. 1). Make sure the Plot checkbox is selected.
Figure 25:
RSMD Trajectory Tool. The RMSD is plotted for the equilibration of ubiquitin.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=0.8\textwidth]{FIGS/rmsd-tool}
}
\end{center}
\end{figure}](img73.gif) |
- 4
- Click the Align button. This will align each frame of the trajectory with respect to the reference frame (in this case, frame 0) to minimize RMSD, by applying only rigid-body translations and rotations. This step is not necessary, but is desirable in most cases, because we are interested only in RMSD that arises from the fluctuations of the structure and not from the displacements and rotations of the molecule as a whole. The result of the alignment can be seen in the OpenGL display.
- 5
- Now, click the RMSD button in the RMSD Trajectory Tool window. The protein RMSD (in Ångstroms) vs. frame number is displayed in a plot (Fig. 25).
- 6
- We will now work with the other trajectory. Load the pulling trajectory into VMD using the files ubiquitin.psf and pulling.dcd. Make sure you load ubiquitin.psf as a new molecule. You can change the names of the molecules by double-clicking on them in the VMD Main menu (see Section 5.1.2).
- 7
- In the RMSD Trajectory Tool window, hit the button Add all to update the list of molecules.
- 8
- Click the Align button, and then click RMSD. The new graph (Fig. 26) displays two RMSD plots vs. time, one for the equilibration trajectory, and the other for the pulling trajectory. The pulling RMSD does not level off and is much higher than the equilibration RMSD, since the protein is stretched in the simulation.
- 9
- Quit VMD.
Figure 26:
RMSD vs. time for the equilibration (blue) and pulling (red) trajectories of ubiquitin.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=0.8\textwidth]{FIGS/rmsd-2}
}
\end{center}
\end{figure}](img75.gif) |
Example of an analysis script
In many cases, one requires special types of trajectory analysis that are tailored for certain needs. The Tcl scripting in VMD provides opportunities for such custom tasks. Users commonly write their own scripts to analyze the features of interest, and a very extensive library of VMD scripts, contributed by many users, is available online, at http://www.ks.uiuc.edu/Research/vmd/script_library/.
We will investigate a very simple exemplary script, distance.tcl, which computes the distance between two atom selections vs. time and also distribution of the distance.
- 1
- Start a new VMD session. Load the ubiquitin equilibration trajectory (files ubiquitin.psf and equilibration.dcd).
- 2
- Open the TkConsole window by selecting Extensions
Tk Console in the VMD Main menu.
- 3
- In the the TkConsole window, load the script into VMD by typing: source distance.tcl (make sure that the file distance.tcl is in the current folder). This will load the procedure defined in distance.tcl into the VMD.
- 4
- One can now invoke the procedure by typing distance in the the TkConsole window. In fact, the correct usage is
distance seltext1 seltext2 N_d f_r_out f_d_out
where seltext1 and seltext2 are the selection texts for the groups of atoms between which the distance is measured, N_d is the number of bins for the distribution, and f_r_out and f_d_out are the file names to where the output distance vs. time and distance distribution will be written.
- 5
- Open the script file distance.tcl with a text editor. You can see that the script does the following:
- Choose atom selections
set sel1 [atomselect top "$seltext1"]
set sel2 [atomselect top "$seltext2"]
- Get the number of frames in the trajectory and assign this value to the variable nf
set nf [molinfo top get numframes]
- Open file specified by the variable f_r_out
set outfile [open $f_r_out w]
- Loop over all frames
for {set i 0} {$i < $nf} {incr i} {
- Write out the frame number and update the selections to the current frame
puts "frame $i of $nf"
$sel1 frame $i
$sel2 frame $i
- Find the center of mass for each selection (com1 and com2 are position vectors)
set com1 [measure center $sel1 weight mass]
set com2 [measure center $sel2 weight mass]
- At each frame i, find the distance by subtracting one vector from the other (command vecsub) and computing the length of the resulting vector (command veclength), assign that value to an array element simdata($i.r), and print a frame-distance entry to a file
set simdata($i.r) [veclength [vecsub $com1 $com2]]
puts $outfile "$i $simdata($i.r)"
}
- Close the file
close $outfile
- The second part of the script is for obtaining the distance distribution. It starts from finding the maximum and minimum values of the distance
set r_min $simdata(0.r)
set r_max $simdata(0.r)
for {set i 0} {$i < $nf} {incr i} {
set r_tmp $simdata($i.r)
if {$r_tmp < $r_min} {set r_min $r_tmp}
if {$r_tmp > $r_max} {set r_max $r_tmp}
}
- The step over the range of distances is chosen based on the number of bins N_d defined in the beginning, and all values for the elements of the distribution array are set to zero
set dr [expr ($r_max - $r_min) /($N_d - 1)]
for {set k 0} {$k < $N_d} {incr k} {
set distribution($k) 0
}
- The distribution is obtained by adding 1 (incr ...) to an array element every time the distance is within the respective bin
for {set i 0} {$i < $nf} {incr i} {
set k [expr int(($simdata($i.r) - $r_min) / $dr)]
incr distribution($k)
}
- Write out the file with the distribution
set outfile [open $f_d_out w]
for {set k 0} {$k < $N_d} {incr k} {
puts $outfile "[expr $r_min + $k * $dr] $distribution($k)"
}
close $outfile
Figure 27:
Distance between a residue and the center of ubiquitin. The distances analyzed are those for residue 76 (black) and residue 10 (green).
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=0.7\textwidth]{FIGS/script-analyze}
}
\end{center}
\end{figure}](img76.gif) |
- 6
- Now run the script by typing in the TkConsole window
distance "protein" "protein and resid 76" 10 res76-r.dat res76-d.dat
This will compute the distance between the center of the protein and center of the terminal residue 76, and write the distance vs. time and its distribution to files res76-r.dat and res76-d.dat.
- 7
- Repeat the same for the protein's residue 10 by typing in the TkConsole window
distance "protein" "protein and resid 10" 10 res10-r.dat res10-d.dat
The data in files produced by the script distance.tcl are in two-column format. Compare the outputs for residue 76 and 10 using your favorite external plotting program (Fig. 27).
Residue 76 is at the protein's C-terminus, which is extended towards the solvent and is quite flexible, while residue 10 is at the surface of the globular part of ubiquitin. The difference in their dynamics with respect to the rest of the protein is immediately obvious when our newly obtained data are plotted (Fig. 27): the distance of residue 76 from the protein's center is substantially greater than that of residue 10, and the distribution of the distance is noticeably wider due to the flexibility of the C-terminus. This is just a simple example of scripting for the analysis of a trajectory. Similar, but usually much more complex, customized scripts are routinely employed by VMD users to perform many kinds of analysis.
- 8
- Quit VMD.
Next: Working with Multiple Molecules
Up: VMD Tutorial
Previous: Scripting in VMD
vmd@ks.uiuc.edu
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2.5...
...er keywords, such as residue, are consistent
with the PDB file.}
\end{minipage}}](img67.gif)
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2.5...
... {Open GL Display} window is active
when using these shortcuts.}
\end{minipage}}](img68.gif)
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2.5...
...fections of the simulation
force-fields can contribute as well.}
\end{minipage}}](img74.gif)
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2.5...
...ared, and $r_{i}(t)$\ is the position of atom $i$\ at time $t$.}
\end{minipage}}](img70.gif)