


Next: MDFF with Domain Restraints
Up: MDFF Tutorial
Previous: A simple MDFF example
Contents
Subsections
MDFF with explicit solvent
In the last section you learned how to set up a simple MDFF simulation
in vacuo. Now you will learn how to set up a similar simulation in
explicit solvent.
Preparing the initial structure
We will start with the structure that was already prepared in the
previous section, i.e., files 1ake-initial-docked_autopsf.psf and 1ake-initial-docked_autopsf.pdb.
- 1
- Start a new VMD session.
- 2
- Load the initial structure you prepared for MDFF in vacuo in the
previous section:
| mol new 1ake-initial-docked_autopsf.psf |
|
| mol addfile 1ake-initial-docked_autopsf.pdb |
|
- 3
- Embed this structure into a water box using the solvate
plugin. In the VMD Main Window, choose Extensions
 Modeling
Add Solvation Box (Fig. 8). Set
the box padding to 20Å for maximum y, 5Å for minimum y, and
10Å for the remainder dimensions, as shown in the figure and click
on Solvate. We would like the target map to fall
completely within the water box, which is why we chose a larger padding
in one of the dimensions. VMD will generate the files solvate.psf
and solvate.pdb, which will be automatically loaded upon
completion of this step. Load the target map (from the previous section)
to visually ensure that the density falls completely within the water
box (Fig. 9).
Modeling
Add Solvation Box (Fig. 8). Set
the box padding to 20Å for maximum y, 5Å for minimum y, and
10Å for the remainder dimensions, as shown in the figure and click
on Solvate. We would like the target map to fall
completely within the water box, which is why we chose a larger padding
in one of the dimensions. VMD will generate the files solvate.psf
and solvate.pdb, which will be automatically loaded upon
completion of this step. Load the target map (from the previous section)
to visually ensure that the density falls completely within the water
box (Fig. 9).
Figure 8:
The VMD Solvate window.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[scale=0.5]{FIGS/solvate}
}
\end{center}
\end{figure}](img23.gif) |
Figure 9:
By loading the target density map you can
visually ensure that the target density falls within the boundaries of
the water box. If part of the target density is outside the water box,
you should adjust the padding accordingly and regenerate the water
box.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[scale=0.4]{FIGS/map-water-box}
}
\end{center}
\end{figure}](img24.gif) |
- 4
- For MD simulations in explicit solvent, it is usually desirable
to have a neutrally charged system. This can be achieved by adding
neutralizing counterions to the simulation system. One can also add
additional ions to mimic in vivo or in vitro conditions. In this
example, we will simply neutralize the system by adding either Na
 or
Cl
or
Cl ions using the autoionize plugin. In the VMD Main Window,
choos Extensions
Modeling
Add Ions
(Fig. 10). Uncheck the button defining the ion
concentration, leaving only neutralization active, as shown in the
figure. Click on Autoionize. VMD will generate the files
ionized.psf and ionized.pdb.
ions using the autoionize plugin. In the VMD Main Window,
choos Extensions
Modeling
Add Ions
(Fig. 10). Uncheck the button defining the ion
concentration, leaving only neutralization active, as shown in the
figure. Click on Autoionize. VMD will generate the files
ionized.psf and ionized.pdb.
Figure 10:
The VMD Autoionize window.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[scale=0.5]{FIGS/autoionize}
}
\end{center}
\end{figure}](img27.gif) |
- 5
- Generate a PDB file containing the per-atom scaling factors
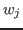 in Equation 1, as in the previous section. By default, water
molecules and ions are not coupled to the target map, i.e., they can
equilibrate freely according to the MD force field and don't experience
additional forces from MDFF:
in Equation 1, as in the previous section. By default, water
molecules and ions are not coupled to the target map, i.e., they can
equilibrate freely according to the MD force field and don't experience
additional forces from MDFF:
| mdff gridpdb -psf ionized.psf -pdb ionized.pdb |
|
| -o ionized-grid.pdb |
|
- 6
- Generate secondary structure restraints as in the previous
section:
| package require ssrestraints |
|
| ssrestraints -psf ionized.psf -pdb ionized.pdb |
|
| -o ionized-extrabonds.txt -hbonds |
|
- 7
- Generate restraints to prevent cis/trans peptide transitions
and chirality errors:
| mol new ionized.psf |
|
| mol addfile ionized.pdb |
|
| cispeptide restrain -o ionized-extrabonds-cispeptide.txt |
|
| chirality restrain -o ionized-extrabonds-chirality.txt |
|
Preparing the density map
As you can see in Fig. 9, we already ensured that the
target density map (blue) is completely within the water box, which is a
requirement for MDFF in explicit solvent. However, the current
implementation of NAMD's gridforces feature also requires that
the entire target map be within the water box, which is clearly not the
case (Fig. 9). An option, gridforcechecksize off or
mgridforcechecksize 0 off if using mgridforce, can be used
to ignore the warning. Additionally, to address this issue directly, we can
trim the target map so that it lies completely within the water box.
- 1
- Trim the map in all dimensions by a few Angstroms
to ensure it will be within the water box during the simulation. The
voltool command provides some features for manipulating volumetric
maps. To trim the target map by 7Å in all dimensions, run:
| voltool trim -i 4ake-target_autopsf-grid.dx -amt 7 7 7 7 |
|
| -o 4ake-target_autopsf-grid-trimmed.dx |
|
- 2
- Visualize in VMD the water box and the newly trimmed map to verify
that it falls completely within the water box. Also ensure that the target
macromolecular volume is contained in the new map. Load the trimmed map
with the command:
| mol new 4ake-target_autopsf-grid-trimmed.dx |
|
Generate NAMD configuration files similarly to the previous section.
- 1
- For MDFF simulations in solvent we need to define periodic
boundary conditions. We also use a different method to calculate
electrostatic interactions that is more appropriate for this kind of
simulation. All of this is taken care of by providing the extra option
-pbc to mdff setup:
| mdff setup -pbc -o adk-solvent -psf ionized.psf |
|
| -pdb ionized.pdb |
|
| -griddx 4ake-target_autopsf-grid-trimmed.dx |
|
| -gridpdb ionized-grid.pdb |
|
| -extrab {ionized-extrabonds.txt ionized-extrabonds-cispeptide.txt |
|
| ionized-extrabonds-chirality.txt} -gscale 0.3 -numsteps 100000 |
|
Note that we requested a simulation twice as long as in the previous
section, since explicit-solvent MDFF simulations typically take longer
to converge.
- 2
- Once again, generate a second NAMD configuration file in which
only energy minimization will be performed with a much higher scaling
factor
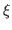 :
:
| mdff setup -pbc -o adk-solvent -psf ionized.psf |
|
| -pdb ionized.pdb |
|
| -griddx 4ake-target_autopsf-grid-trimmed.dx |
|
| -gridpdb ionized-grid.pdb |
|
| -extrab ionized-extrabonds.txt ionized-extrabonds-cispeptide.txt |
|
| ionized-extrabonds-chirality.txt} -gscale 10 |
|
| -minsteps 2000 -numsteps 0 -step 2 |
|
- 3
- Quit VMD.
- 4
- Run NAMD using the configuration files generated by VMD, i.e.,
run the following commands in a terminal (or submit them to a cluster):
| namd2 adk-solvent-step1.namd > adk-solvent-step1.log |
|
| namd2 adk-solvent-step2.namd > adk-solvent-step2.log |
|
This step should take about 40 minutes on a cluster with 48
processors. If you don't want to wait, you can proceed to the next
step and use the provided trajectory files, as explained in the next
section.
The resulting trajectories will be saved to files adk-solvent-step1.dcd and adk-solvent-step2.dcd. If you want to
continue working through the tutorial before the simulations are
complete, you can use the provided trajectory files adk-step1-result.dcd and adk-step2-result.dcd instead. Once
again, please note that due to the stochastic nature of molecular
dynamics simulations it is expected that the trajectories obtained will
differ from the ones provided. As in the previous section, load the
resulting trajectory, as well as the target structure, and repeat the
analysis of the RMSD ad CCC. Did the use of explicit solvent improve the
MDFF results in this particular case?
Next: MDFF with Domain Restraints
Up: MDFF Tutorial
Previous: A simple MDFF example
Contents
school@ks.uiuc.edu