

Next: Minimization with new parameters
Up: Parameterization Tutorial
Previous: Developing Topology and Parameter
Subsections
Parameter generation using SPARTAN (Optional)
This section relies on the program Spartan for semi-empirical parameter generation.
If you do not have Spartan available to you, read the section in its entirety, but without
doing any of the specific steps. You will not need anything produced in this section to
complete the tutorial.
As you know by now, the glutamine-cysteine linkage is defined as
a new residue in the CHARMM27 topology file with the name CYG. To actually
determine the optimum parameter values for equilibrium bond lengths, angles, and to determine
the energetic barriers associated with them, we need to use quantum chemistry. Here, we will use
SPARTAN to generate these values. For the purposes of this tutorial, we will calculate the values
semi-empirically; for a true parameterization in accordance with the CHARMM22/CHARMM27 force fields,
we would need to do full ab-initio calculations with the Hartree-Fock 6-31G* basis set. These types
of calculations would take too long to complete in one afternoon. To compute more rigorous quantum
chemistry calculations on small molecules, one often uses parallelizable quantum chemistry packages,
e.g. GAMESS (freely available, highly recommended) or Gaussian.
For this exercise, we also need to use a highly truncated form
of the residue because of computational restrictions on the number of atoms one
can feasibly calculate with quantum chemistry packages. Presently, the maximum number
of atoms one can expect to realistically compute with high level quantum chemistry is
approximately 120 atoms. You will likely find that attempting to do larger systems will
take a prohibitively long time to compute. When developing the new parameters
for this nonstandard residue, we will neglect any solvent effects and consider the molecule to
be in the gas phase. The truncated form of CYG that we will be working with has the following formula:
CH3-S-CO-CH3 and its IUPAC name is methyl thioethanoate.
To start Spartan, open a terminal and type spartan.
When you launch Spartan, the first thing you see is a large green screen. This is the main Spartan window.
If you look at the top of your display, you will see that Spartan has a menu to the right of the little blue apple; this is
the main Spartan menu. You will also see a small box with symbols appearing somewhere within
your main Spartan window. If you like, you can move the box out of the way by clicking on the bar at the top
of the small window and dragging it to a different location.
You can build a new molecule in Spartan by choosing (from the main Spartan menu):
- File
 New
New
Figure 5:
The Spartan Program
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[scale=0.5]{FIGS/spartan}
}
\end{center}
\end{figure}](img37.gif) |
The Model Kit window should appear. You can build
molecules by selecting atom (and bond) types in the Model Kit window, and then placing
them by clicking in the main Spartan window. Atoms can be
connected by clicking on the open valences. Now we will start building the truncated form of CYG:
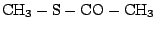 . All the bonds are single except for
the double bond between the carbon and oxygen (a carbonyl group!). If you make a mistake, you
can easily fix it by selecting Edit Undo. To build the truncated molecule:
. All the bonds are single except for
the double bond between the carbon and oxygen (a carbonyl group!). If you make a mistake, you
can easily fix it by selecting Edit Undo. To build the truncated molecule:
- In the Model Kit window, select a methyl group by clicking on it the sp3 carbon option. The sp3 carbon is the
carbon with 4 single bonds sticking out of it.
- Now click anywhere in the Main Spartan Window, and see the carbon appear with 4 open valences.
- Return to the Model Kit window and select the sulfur with two open valances (i.e. the sulfur
with two single bonds sticking out of it).
- Go back to the Main Spartan Window and click on one of the open valences of the sp3 carbon. The sulfur
group should appear!
- Again, return to the Model Kit window and in the Groups dropdown menu, select Carbonyl.
A picture of a carbonyl group, i.e. a carbon with a double bond to an oxygen, should appear in the Model Kit
window.
- Now return again to the Main Spartan Window and click on the open valence of the sulfur. The
carbonyl group should now be attached.
- Finally, in the Model Kit window, select the another sp3 carbon group
- Now add this last carbon to your existing molecule by clicking on the open valence coming off of the
carbonyl group.
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2...
...anslate the molecule, and scroll wheel to scale the molecule.}
\end{minipage} }](img39.gif)
Turn on atom labels by choosing Model: Labels from the main Spartan menu at the top
of your display. When you have done it correctly, you should have something very similar to Fig. 6.
Figure 6:
Building CYG in Spartan
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[scale=0.5]{FIGS/spart1new}
}
\end{center}
\end{figure}](img40.gif) |
Close the Model Kit window by clicking on the red button in the top left corner of the window.
Once you close it, you will see explicit hydrogens appear on any open valences.
Now minimize the structure by selecting Build Minimize from the main
Spartan menu. Once it's done, you should see something identical to Fig. 7.
Figure 7:
CYG after minimization in Spartan
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[scale=0.5]{FIGS/spart2new}
}
\end{center}
\end{figure}](img41.gif) |
Everything is ready now for a semiempirical quantum mechanics
calculation. Now you will optimize the geometry of the structure, calculate electrostatic
potential (ESP) charges and vibrational frequencies.
To do this, from the main Spartan menu, choose Setup Calculations and set the following options:
- Calculate: Equilibrium Geometry at Ground state
- with: Semi-Empirical PM3
- Start from: Initial Geometry
- Subject to: Symmetry
- Compute: IR
- Print: Atomic Charges, Vibrational Modes
- Options: check Converge
It should look identical to Fig. 8. When it does, click OK.
Figure 8:
Setting up the energy calculation.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[scale=0.5]{FIGS/fig8}
}
\end{center}
\end{figure}](img42.gif) |
You can submit the job and view its output by choosing:
- Click on Setup Submit. Choose a name for the job save the job files to the directory you're working in.
- The job will take a couple of minutes to complete. A window will appear indicating the completion of the job
- Then go to Display Output
The output file lists details about the method used, and lists the Cartesian coordinates of the atoms.
You can measure atomic distances and bond angles by using the Geometry pulldown.
Now, we are going to display the vibrational modes. The
modes can be viewed through the animation part of the program.
Click: Display Spectra; when you check the boxes you
can watch individual vibrational modes. Try increasing the amplitude to see what happens; you will
find that the motion of the vibrations become more exaggerated as you increase their amplitudes. Note that
the highest frequencies belong to the hydrogens coming off the methyl groups!
Figure 9:
Vibrations Window
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[scale=0.5]{FIGS/fig9}
}
\end{center}
\end{figure}](img44.gif) |
Figure:
Thioester linkage found in 1ODV.pdb, the photoactive yellow protein.
|
|
A systematic calculation of the force constants for the
bond stretching and angle bending motion can be obtained from the
ab initio calculations [10], but is beyond the scope of
this exercise.
To complete the exercise, be sure you toggle off any selected vibrations. In order to proceed to the
next steps, the molecule should no longer be moving! When this is the case, close the Spectra window.
Now we are going to investigate the partial charges of the atoms. First, we are going to
display a surface around the atoms, then we will map the ESP charges of the molecule onto the surface we generated.
But before we do this, let's check out the numerical values of the Mulliken charges and
ESP charges and compare the difference between them. This can be achieved by doing the following:
- Select Model Ball and Wire
- Select Model Configure In the Configure window, select Mulliken first and later, Electrostatic Charges.
Generally you will see that the partial charge values derived from
an ESP calculation are higher than the Mulliken charges. ESP
charges are calculated to reproduce the electrostatic potential
around an atom, whereas the Mulliken charges are derived from the
electron occupancy of orbitals.
When you are finished investigating the charges, close the Configure window.
You can display a potential surface by:
- Clicking on Setup Surfaces.
- In the new window, select Add.
- Then in the new "Add Surface" window, select Surface density and Property potential and
click OK.
- Submit the surface calculation with Setup Submit.
- When the job completes, you can view the surface with Display Surfaces. In the new "Surfaces" window, you can toggle the surface on and off by checking the box next to density.
Figure 11:
Surface of molecule with ESP charges
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[scale=0.5]{FIGS/surface}
}
\end{center}
\end{figure}](img48.gif) |
Be sure to toggle the surface off to proceed to the next part of the exercise and close the Surface window.
Now is a good time to discuss the determination of the partial charges appearing in the topology
file. Two widely used force fields are CHARMM and AMBER - and they both use different
protocols to develop the partial atomic charges.
CHARMM starts with the Mullikin charges and then strategically places a water molecule near any
polar group. CHARMM's charges are refined so that they best reproduce the interaction energy
between the water and the polar group. To recreate these values, the partial charges of the
polar group are then altered until this interaction energy is accurately replicated with the MD
simulations. Several concrete examples of this charge-fitting strategy are worked out in Ref. [7].
AMBER, on the other hand, begins with the ESP charges and then uses a program they distribute called
RESP (for Restricted ESP). This charge-fitting scheme replicates the electrostatic potential at a distance out
from the nucleus (much like the surface you built in the previous exercise) with only the atom centers having
charge. Of course, the electrostatic surface generated using this method is often degenerate; meaning there could be more
than one set of charges that would reproduce the potential surface. To resolve this issue, it is best
to further restrict your potential surface by supplying it an initial set of charges for the program's iterations, or
further restrictions derived from chemical insight.
However they are generated, the importance of a good set of partial charges cannot be overstated. Much of the
energy (and indirectly then, the behavior) of the system will be calculated using these values.
The next exercise will be to calculate the energy of rotation around the H1-C1-S1-C2 dihedral.
In Spartan, this is called an Energy Profile calculation (there are other names for the same task,
such as conformational sampling, coordinate drive, or dihedral search).
It is essential that you follow the next set of directions as closely as you can. There are a lot of steps and if you miss
one, it is unlikely that you will successfully generate the energy profile we seek!
First we need to identify which dihedral angle we want to investigate. To do this:
- Change the labels back to the atom name so
you are sure you pick the correct atoms by selecting Model Configure Labels from the main Spartan menu
- From the main Spartan menu, select Geometry Measure Dihedral.
- Now return back to the main Spartan window and click on the four atoms of interest in this order: H1-C1-S1-C2
Figure 12:
Dihedral selection
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[scale=0.5]{FIGS/spart4new}
}
\end{center}
\end{figure}](img50.gif) |
On the very bottom of the main Spartan window, there is a bar with information. Right now, the bar should
list the dihedral angle you have chosen, as well as the angle of the dihedral specified in degrees. Please
notice that at the very right hand side of this bar (in other words, the lower right hand corner of the main
Spartan window), there is a picture of a small lock. We will use it shortly.
- From the main Spartan menu, select Display Properties. A Molecule Properties window will appear.
- Now return to the main Spartan window. Click on the small lock symbol in the lower right corner to constrain the dihedral.
You will notice that the Properties Window changes to display Constraint Properties for the dihedral we are interested in.
- Now in the Constraint Properties window, change the values to:
- Value: 0 To: 360
- Steps: 12
- Select: Setup Calculations from the main Spartan menu.
- In the Calculations window, select Calculate: Energy Profile at Ground state,
with: Semi-empirical PM3, Subject to: Symmetry, Print: Atomic Charges,
check Converge. All of the options under Compute: UV/VIS, IR, and NMR
should NOT be checked. Then hit OK.
- Select Setup Submit from the main Spartan menu to submit the job.
Once the calculation is complete (it takes a couple of minutes to run), you can plot the Energy Profile. You may notice that now
there are two copies of the molecule in the main Spartan window. In the next series of steps
we are going to select and hide the old molecule, and then select the new molecule,
as it now has all the different torsion angles loaded into a sequence of coordinate
files named P1, P2, P3, ..., P13. To do this:
- Click on the old molcule (you can tell which molecule you have selected by the name that appears on the bar at the bottom of the main Spartan window.
- Select Model Hide . The old molecule should disappear.
- Click on the new molecule (named P1).
Once you have selected the new molecule, we can plot the Energy Profile by the following steps:
- In the main Spartan menu, select Display Spreadsheet.
- In the new window that appears, click on Add.
- In the Add Column window, click on E, make sure the units of energy are kcal/mol, and then drag it over to the second column in the spreadsheet window.
- Close the Add Column window.
- In the main Spartan menu, select Display Plots. In the Add Column window, keep the X axis set to Molecule, and select the Y axis to be E
(kcal/mol), then click OK.
Figure 13:
Dihedral energy profile results.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[scale=0.5]{FIGS/fig13}
}
\end{center}
\end{figure}](img51.gif) |
Spartan automatically superimposes the plot of the energy profile on top of the molecule. If you click on the ''play forward" button on the bottom bar in
the main window, you can watch the torsion angle change and simultaneously see the corresponding point on the plot. See Fig. 13.
If you were not successful in creating the energy drive plot, you may need to close Spartan completely and rebuild
the truncated molecule; then repeat all the steps again carefully.
When you are finished with the exercise, please quit Spartan.
Next: Minimization with new parameters
Up: Parameterization Tutorial
Previous: Developing Topology and Parameter
tutorial@ks.uiuc.edu
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2...
...(as found in Section 5.2)? Why might the values be different?}
\end{minipage} }](img43.gif)
![% latex2html id marker 1362
\framebox[\textwidth]{
\begin{minipage}{.2\textwid...
...For general
thioester linkage information, see \cite{ZACH82}.}
\end{minipage} }](img45.gif)
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2...
...topology file in Section 5.1?
Which set of values is closer?}
\end{minipage} }](img47.gif)
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2...
... the color scheme on the surface fit the ESP partial charges?}
\end{minipage} }](img49.gif)
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2...
...n from the parameter file in Matlab to do a full comparison. }
\end{minipage} }](img52.gif)
![\includegraphics[width=3.0in]{FIGS/thio}](img46.gif)