

Next: Analysis of Results
Up: Steered Molecular Dynamics
Previous: Constant Velocity Pulling
Subsections
Now, you will carry out an SMD simulation that applies a constant force. In this case, the C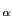 atom of
the first residue is again kept fixed, but the SMD atom C of the last residue now experiences
a constant force in the direction defined by the vector that links both atoms, the fixed and the pulled one.
Note that in this case there is no dummy atom or virtual spring.
atom of
the first residue is again kept fixed, but the SMD atom C of the last residue now experiences
a constant force in the direction defined by the vector that links both atoms, the fixed and the pulled one.
Note that in this case there is no dummy atom or virtual spring.
Again, NAMD uses a column of a pdb file to determine which atoms will be fixed and which atoms will be pulled. In addition, another three columns are used to specify the direction of the constant force that will be applied to the SMD atom. Your first task is to build this file.
- 1
- Navigate back to the namd-tutorial-files/ directory by typing in a Terminal window cd ..
- 2
- In your opened session of VMD, choose the File
 New Molecule... menu item and using
the Browse... and the Load buttons load the file common/ubq.psf located in the common directory, and close the
Molecule File Browser window.
New Molecule... menu item and using
the Browse... and the Load buttons load the file common/ubq.psf located in the common directory, and close the
Molecule File Browser window.
- 3
- Using the mouse select the molecule in the VMD Main window and then choose
the Load Data Into Molecule... menu item. Again, using the Browse... and the Load
buttons load the file ubq_ww_eq.pdb located in the common directory. Close the Molecule File Browser.
- 4
- As you did in the previous section, define your fixed atom by typing the following commands
in the VMD TkCon window:
| set allatoms [atomselect top all] |
|
| $allatoms set beta 0 |
|
| set fixedatom [atomselect top "resid 1 and name CA"] |
|
| $fixedatom set beta 1 |
|
$allatoms set occupancy 0
|
|
- 5
- The occupancy of the SMD atom will contain the force applied to it. Type
| set smdatom [atomselect top "resid 76 and name CA"] |
|
$smdatom set occupancy 11.54
|
|
You have now set the force to 11.54 kcal/mol/Å by entering the value into the occupancy field. It is equivalent to 800 pN.
- 6
- The direction of the force will be specified in the coordinates of the SMD atom. Therefore, you have to write
the normal vector in the following way:
$smdatom set x 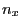 |
|
$smdatom set y 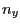 |
|
$smdatom set z 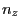
|
|
where , , and have to be replaced by the appropriate values you already calculated above (in our example 0.352, 0.402, and 0.845). Since the VMD OpenGL Display interprets the numbers just entered as coordinates of the SMD atom, it displays now an inaccurate protein structure. This is OK and we apologize for the poor appearance.
- 7
- Now, save the coordinates into a file by typing $allatoms writepdb common/ubq_ww_eq2.ref
- 8
- Delete the current molecule by choosing the Molecule Delete Molecule menu item and keep VMD opened.
As you did before, in the next step you will modify the configuration file in order to set up your constant
force simulation.
- 1
- Open a new Terminal window, and change the directory to namd-tutorial-files/.
- 2
- Copy the file common
 sample.conf to the directory 3-2-pullcf and rename it by typing copy commonsample.conf 3-2-pullcfubq_ww_pcf.conf.
sample.conf to the directory 3-2-pullcf and rename it by typing copy commonsample.conf 3-2-pullcfubq_ww_pcf.conf.
- 3
- Now, open the configuration file 3-2-pullcfubq_ww_pcf.conf using WordPad.
- 4
- As a job description you can write
# N-C-Termini Constant Force Pulling
- 5
- In the Adjustable Parameters section you need to change:
| structure mypsf.psf |
|
structure ../common/ubq.psf |
| coordinates mypdb.pdb |
|
coordinates ../common/ubq_ww_eq.pdb |
| outputName myoutput |
|
outputName ubq_ww_pcf |
In this way you are using the equilibrated protein without water in your simulation. The output
files of your simulation will have the prefix ubq_ww_pcf in their names.
- 6
- In the Input section change:
parameters par_all27_prot_lipid.inp
|
|
parameters ../common/par_all27_prot_lipid.inp
- 7
- As before, switch off the Constant Temperature Control by changing:
| langevin on |
|
langevin off |
- 8
- Enable the Fixed Atoms Constraint by changing the following lines
| if {0} { |
|
if {1} { |
| fixedAtomsFile myfixedatoms.pdb |
|
fixedAtomsFile |
| |
|
../common/ubq_ww_eq2.ref |
NAMD will keep fixed the atoms which have a B value of 1 in the file ../common/ubq_ww_eq2.ref.
- 9
- In the Extra Parameters section add the following lines:
| constantforce |
yes |
| consforcefile |
../common/ubq_ww_eq2.ref |
NAMD will apply a constant force to the atoms that have occupancy different from 0. The force
is calculated from the file as
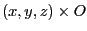 , where
, where 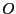 is the value of the occupancy column.
is the value of the occupancy column.
- 10
- Finally, in the Execution Script section of your configuration file be sure that the minimization is disabled and change the number of time steps your simulation will run by replacing:
| run 50000 |
|
run 20000 |
This is equivalent to 40 ps.
- 11
- Your second configuration file is done. SAVE IT (in text-only format) and close the text editor.
Now, all the files you need to launch your second simulation are ready. You should have a file called ubq_ww_pcf.conf
in the 3-2-pullcf directory and files:
- ubq.psf
- ubq_ww_eq.pdb
- ubq_ww_eq2.ref
- par_all27_prot_lipid.inp
in the common directory. In case that you have not generated these files you can use prepared files available at
directories 3-2-pullcf/example-output and common/example-output.
- 1
- In a Terminal window, change to directory 3-2-pullcf/ to run the simulation.
Note that the output file has no extra information in this case,
since the applied force is now constant.
Next: Analysis of Results
Up: Steered Molecular Dynamics
Previous: Constant Velocity Pulling
namd@ks.uiuc.edu
