To get started with RMS fitting and alignment, type vmdrmsd from the command line. Alternatively, you can source the script called rmsd.tcl, which should be located in the scripts/vmd subdirectory of your VMD installation. For example, if VMD is installed in /usr/local/lib/vmd, type the following from the command line: source /usr/local/lib/vmd/scripts/vmd/rmsd.tcl. You should now have two new menus, titled RMSD calculator and RMS Alignment (kindly provided by Alexander Balaeff from the Theoretical Biophysics Group). We'll describe the RMSD calculator first.
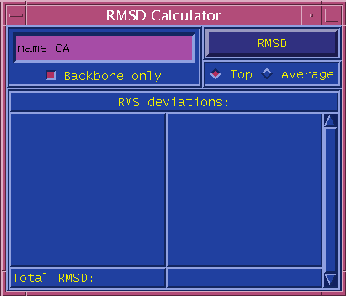
The RMSD Calculator menu is used to calculate RMS distances between molecules.
The upper left corner of the menu is where you specify which atoms are to be used in the calculation. In the input field, type the atom selection text just as you would in the Graphics form. The checkbox below the input field entitled "Backbone only" restricts whatever atom selection you typed to just the backbone atoms of the selection; in effect, it adds "and backbone" to the atom selection text.
The upper right corner of the menu has a button labeled "RMSD". Its effect depends on which of the two radio buttons, "Top" or "Average", is pressed down. If "Top" is pressed, VMD calculates the RMS distance between the top molecule (which is usually the last molecule loaded) and every other molecule. If "Average" is pressed, VMD first computes the average x, y, z coordinates of the selected atoms in each molecule, then computes the RMS distance of each molecule from that average structure.
Results of the RMS calculations for each molecule are shown in the browser in the bottom half of the menu. Note that this list is not updated until you presse the RMSD button, so the effects of loading/deleting molecules will not be immediately reflected. The "Total RMSD" label at the bottom of the menu shows the average RMSD for all molecules listed.
The RMS Alignment menu fits molecules based on selected groups of atoms. Whereas the RMSD Calculator finds the RMS distance between molecules without disturbing their coordinates, the RMS Alignment menu actually moves molecules to new positions.
This menu is quite simple: Enter an atom selection in the input field, and press Align to align the molecules based on the atoms in that selection. If you recompute the RMSD between molecules in the RMSD Calculator menu, you will probably find that the values are different; this is because the calculation is made based on the current positions of the atoms.