From: MEHRAN MB (mb.mehran1_at_gmail.com)
Date: Tue Jan 27 2015 - 17:09:19 CST
Dear NAMD experts,
I am trying to compare free energy of forming helix structure for two coils
with 28 residues in length. Both are identical except, in one all GLU are
substituded by ALA. Indeed, I want to measure these residue contribution in
forming helix, and I need to calculate Free energy with high precision.
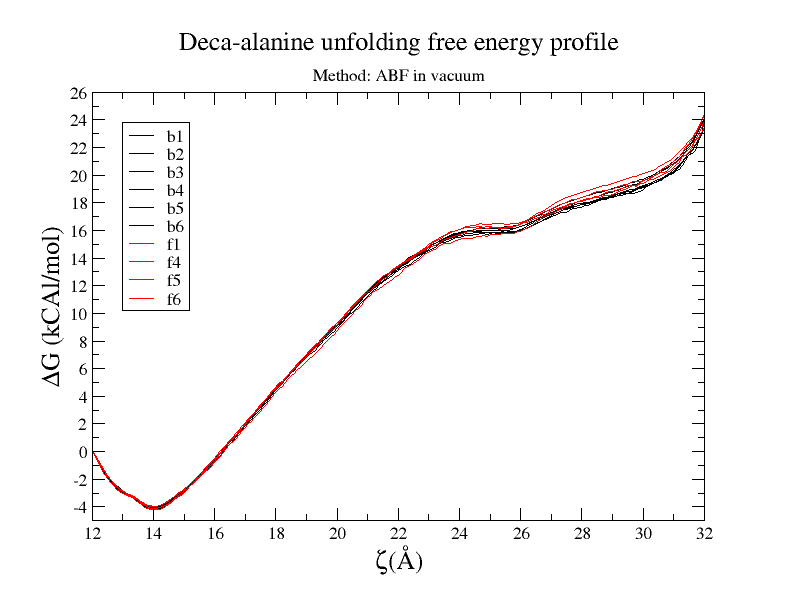
I tried ABF method and it works great for deca-alanine in vacuum. I run
multiple simulations and all gave me pretty consistence results, both in
curve shape and final value. free-energy vs reaction-coordinates result is
provided. (Also when I solvate them in water I could not get good result)
[image: Inline image 1]
However trying ABF on my 28 res polypeptide in vacuum, set boundary on
end-to-end distance to vary from 40A to 80A and #samples within 40000 to
200,000; I have seen pretty inconsistency in free energy curve and final
value. Since there are few charged residues I thought it might trapped in a
non-equilibrium state.
I divided the ABF process to smaller windows. I tried first small window,
end-to-end distance varying from 40A to 45A and sampling from 100,000 to
200,000, and I am still getting pretty inconsistent result ( from 5 to 10
Kcal/mol ) for this window.
So I believe I have enough sampling but it does not necessary mean that It
has been sampled in equilibrium state. So How I can make sure that I am
sampling in *quasi*-*equilibrium* process ?
Next step would be measuring this helix unfolding Free energy in solution
since I doubt I can say anything about the free energy in solution from
vacuum simulation results. (I might be wrong, )
any advice help to improve ABF results or suggestion about other methods
will be appreciated,
Best,
Mehran
UOttawa
here is simulation details:
0.5 timesteps
rigidbond none
0.1 bin width
1000 fullsampling
This archive was generated by hypermail 2.1.6 : Tue Dec 27 2016 - 23:20:51 CST